“Artificial intelligence in risk prediction and mendelian diseases diagnosis”: Today, I want to give a talk about application of AI in risk prediction and disease diagnosis with deep sequencing and deep phenotyping. As the technique growth, we have already had the ability to generate deep sequencing data within short time, however, deep phenotyping is not easy work which limited the development of genetic association study. Actually, we can apply ** molecular phenotypes ** to replace “clinical or external phenotypes”.
Artificial intelligence in risk prediction and diseases diagnosis
How to design and conduct a biomedical research in MCRI
Today, I just submitted the letter of intent for the interal grant application in Marshfiled Clinic Research Institute as the PI. The proposal I submitted this year named: Deep Learning Prediction of Chemotherapy Response using Multi-Omics Features. Same with other grant, the first step of the application is submit letter of intent. You need prepare the title, PI/co-PIs, research abstract including background, hypothesis, aims, strategy and significance. You don’t need to get signature from all the co-PIs in this step. Usually the research abstract should be less than 500 words. You should submit 2 potential reviewer to review your abstract.
After you prepared these materials, you need to submit the information to “Intent to submit” from Research Hub (RH). Just input the information one by one. The whole process usually takes 5-10 mins. and then you will receive the confirmation email to let you know you can prepare the proposal now. usually, the proposal should be less than 6 page. When you prepare the “letter of intent”, you need discuss carefully with all the co-PIs and make the final version and receive confirmation to agree to submission. It become hard to receive even internal grant since this round, MCRI will only support 3 proposal while more than 30 proposals were submitted. Please sure leave enough time for you co-PIs to give comments and approval since some of them are quite busy. I suggest to ask for comments at least 7 days advanced.
This year, I submitted a pure computational proposal without any biology and clincial/genomic data collection. I proposed a novel method to a public dataset since the internal grant quota is only 65,000. It is little better than UW-Madison which is 50,000$. Hope this proposal can be funded.
Genome-wide cell-free DNA fragmentation in patients with cancer
Today, I will give a talk about “Genome-wide cell-free DNA fragmentation in patients with cancer” in MCRI postdoc club meeting. The paper was published in Nature recently. See the slice here or download here for more details.
cfDNA fragmentation in human cancers
Cell-free DNA in the blood provides a non-invasive diagnostic avenue for patients with cancer1. However, characteristics of the origins and molecular features of cell-free DNA are poorly understood. Here we developed an approach to evaluate fragmentation patterns of cell-free DNA across the genome, and found that profiles of healthy individuals reflected nucleosomal patterns of white blood cells, whereas patients with cancer had altered fragmentation profiles. We used this method to analyse the fragmentation profiles of 236 patients with breast, colorectal, lung, ovarian, pancreatic, gastric or bile duct cancer and 245 healthy individuals. A machine learning model that incorporated genome-wide fragmentation features had sensitivities of detection ranging from 57% to more than 99% among the seven cancer types at 98% specificity, with an overall area under the curve value of 0.94. Fragmentation profiles could be used to identify the tissue of origin of the cancers to a limited number of sites in 75% of cases. Combining our approach with mutation-based cell-free DNA analyses detected 91% of patients with cancer. The results of these analyses highlight important properties of cell-free DNA and provide a proof-of-principle approach for the screening, early detection and monitoring of human cancer.
Cell-Free DNA Project and Adavance
- 09/29/2019: Shanghai Changhai Hospital: Prospective Estimation on Stool-DNA Screening of Colorectal Cancer
- 09/28/2019: ESMO, GRAIL (Dr. Geoffrey R Oxnard) anounce cfDNA methylation for tissue-of-origin prediction in 20+ cancers
Cell-Free DNA General Background
- Wei Chen, Noninvasive chimeric DNA profiling identifies tumor-originated HBV integrants contributing to viral antigen expression in liver cancer
- Pan et al “Brain Tumor Mutations Detected in Cerebral Spinal Fluid”
- Schilling and Rehli “Global, comparative analysis of tissue-specificpromoter CpG methylation”
- Jahr et al “DNA Fragments in the Blood Plasma of CancerPatients: Quantitations and Evidence for Their Origin from Apoptotic and Necrotic Cells”
- Van der Vaart and Pretorius “Circulating DNAIts Origin and Fluctuation”
- Stroun et al “The Origin and Mechanism of Circulating DNA”
- Gravina et al “The dark side of circulating nucleic acids”
- Sun et al “Plasma DNA tissue mapping by genome-wide methylationsequencing for noninvasive prenatal, cancer, and transplantation assessments”
- Wyatt et al “Concordance of CirculatingTumor DNA and MatchedMetastatic Tissue Biopsy in Prostate Cancer”
- Ma et al “Cell-Free DNA Provides a Good Representationof the Tumor Genome Despite Its Biased Fragmentation Patterns”
Cell-Free DNA and Methylation
- Lehman-Werman et al “Identification of tissue-specific celldeath using methylation patterns of circulating DNA”
- Synder et al “Cell-free DNA Comprises an In Vivo NucleosomeFootprint that Informs Its Tissues-Of-Origin”
- Jensen et al “Whole genome bisulfite sequencing ofcell-free DNA and its cellular contributorsuncovers placenta hypomethylated domains”
- Guo et al “Identification of methylation haplotypeblocks aids in deconvolution of heterogeneoustissue samples and tumortissue-of-origin mapping from plasma DNA”
- Kessler et al “CpG methylation differences betweenneurons and glia are highlyconserved from mouse to human”
- Luck et al “A Stochastic Model for the Formation of SpatialMethylation Patterns”
- Doherty and Couldrey “Exploring genome wide bisulfite sequencing for DNA methylation analysis in livestock: a technical assessment”
- Intro methylation review: “DNA methylation: a form ofepigenetic control of gene expression”
- Koch et al “Analysis of DNA methylation in cancer: location revisited”
- Smith and Meissner “DNA methylation: roles in mammalian development”
Non-CpG Methylation
- Xu and Corces “Nascent DNA methylome mapping reveals inheritance of hemimethylation at CTCF/cohesin sites”
Deconvolution algorithms
- Houseman et al “Reference-freedeconvolution of DNA methylationdata and mediation by cellcomposition effects”
- Rahmani et al “Sparse PCA Corrects forCell-Type Heterogeneity in Epigenome-WideAssociation Studies”
- Gond and Szustakowski “DeconRNASeq: a statistical framework fordeconvolution of heterogeneoustissue samples based on mRNA-Seq data”
WGBS Algorithms
- Michael Scherer, Quantitative comparison of within-sample heterogeneity scores for DNA methylation data
- Ziller et al “Coverage recommendations for methylation analysis by whole genome bisulfite sequencing”
- Merkel et al “gemBS - high throughput processing for DNA methylation data from Bisulfite Sequencing”
- Lee et al “An integrative approach for efficientanalysis of whole genome bisulfite sequencing data”
- Kunde-Ramamoorty et al “Comparison and quantitativeverification of mapping algorithmsfor whole-genome bisulfite sequencing”
- Xi and Li “BSMAP: whole genome bisulfitesequence MAPping program”
- Guo et al “BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data”
- Krueger and Andrews “Bismark: aflexible aligner and methylationcaller for Bisulfite-Seq applications”
- Feng et al “Disease prediction by cell-free DNA methylation”
Methylation pipeline and analysis tools
- MethylMix 2.0: an R package for identifying DNA methylation genes
- RnBeads 2.0: comprehensive analysis of DNA methylation data
- Bismark:A tool to map bisulfite converted sequence reads and determine cytosine methylation states
-
MethPipe: a computational pipeline for analyzing bisulfite sequencing data
-
see the slice for the presentation. Also you can download here for more details..
-
September 24, 2019 07:00 AM Eastern Daylight Time: CAMBRIDGE, Mass. & SAN CARLOS, Calif.–(BUSINESS WIRE)–Foundation Medicine, Inc. and Natera, Inc. (NASDAQ: NTRA) today announced a partnership to develop and commercialize personalized circulating tumor DNA (ctDNA) monitoring assays, for use by biopharmaceutical and clinical customers who order FoundationOne®CDx. The initial focus of the partnership will be to enable ctDNA monitoring in biopharmaceutical trials in 2020 to establish the clinical utility for these novel assays. Following these studies, a monitoring product will be made available to clinical customers.
- All the figures are only used for non-profit education. reminding me if infrigement happens
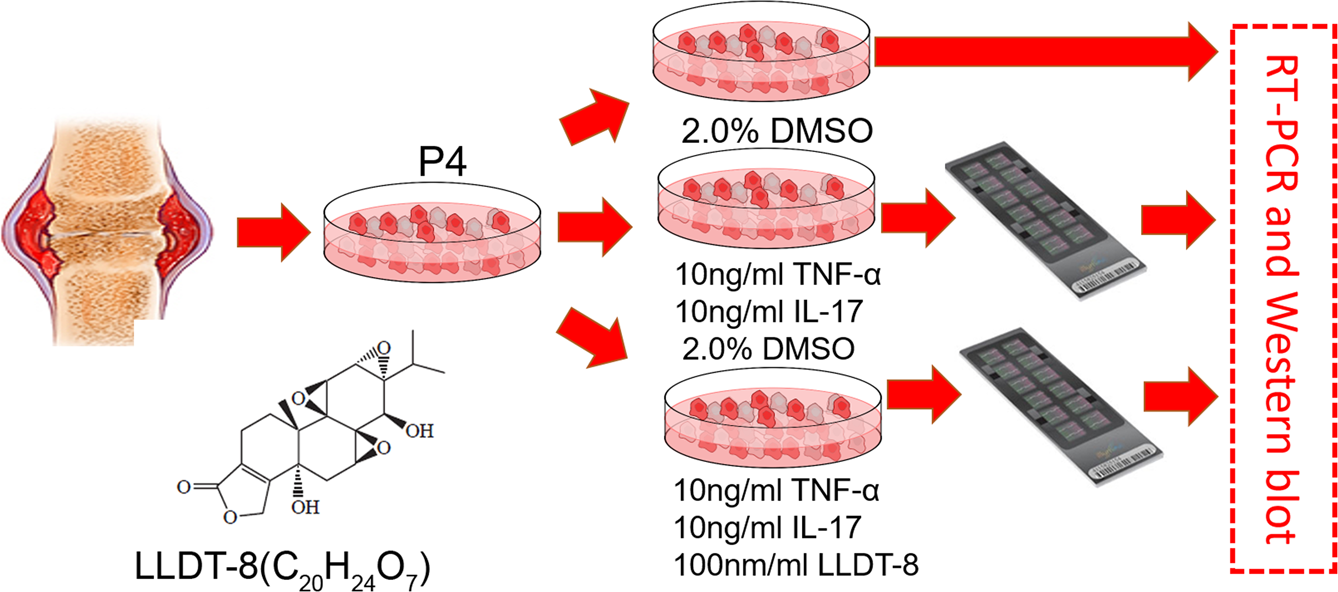
(5R)-5-Hydroxytriptolide and Epigenetics of Rheumatoid Arthritis
Recently, our project collaborated with Dr. Dongyi He in effects of (5R)-5-Hydroxytriptolide to epigenetic changes of LFS from RA was accepted by Scientfic Reports
Tripterygium is a traditional Chinese medicine that has widely been used in the treatment of rheumatic disease. (5R)-5-hydroxytriptolide (LLDT-8) is an extracted compound from Tripterygium, which has been shown to have lower cytotoxicity and relatively higher immunosuppressive activity when compared to Tripterygium. However, our understanding of LLDT-8-induced epigenomic impact and overall regulatory changes in key cell types remains limited. Doing so will provide critically important mechanistic information about how LLDT-8 wields its immunosuppressive activity. The purpose of this study was to assess the effects of LLDT-8 on transcriptome including mRNAs and long non-coding RNA (lncRNAs) in rheumatoid arthritis (RA) fibroblast-like synoviocytes (FLS) by a custom genome-wide microarray assay. Significant differential expressed genes were validated by QPCR. Our work shows that 394 genes (281 down- and 113 up-regulated) were significantly differentially expressed in FLS responding to the treatment of LLDT-8. KEGG pathway analysis showed 20 pathways were significantly enriched and the most significantly enriched pathways were relevant to Immune reaction, including cytokine-cytokine receptor interaction (P = 4.61 × 10−13), chemokine signaling pathway (P = 1.01 × 10−5) and TNF signaling pathway (P = 2.79 × 10−4). Furthermore, we identified 618 highly negatively correlated lncRNA-mRNA pairs from the selected significantly differential lncRNA and mRNA including 27 cis-regulated and 591 trans-regulated lncRNA-mRNAs modules. KEGG and GO based function analysis to differential lncRNA also shown the enrichment of immune response. Finally, lncRNA-transcription factor (TF) and lncRNA-TF-mRNA co-expression network were constructed with high specific network characteristics, indicating LLDT-8 would influence the expression network within the whole FLS cells. The results indicated that the LLDT-8 would mainly influence the FLS cells systemically and specially in the process of immune related pathways.
…
The aim of this study was designed to identify the mechanism of immuno-impressive function of LLDT-8 and identify potential ncRNA targets of LLDT-8. We applied TNF-α and IL-17 to induce an inflammatory status in the cells so that we can observe the LLDT-8 effect easily compared with treatment to normal FLS cell directly. We then assessed the effects of LLDT-8 on the regulation of gene expression on mRNAs and long non-coding RNAs in FLS isolated from RA patients. Agilent Human lncRNA (4 × 180 K, Design ID: 062918) was applied in present study to provide genome-wide ncRNA and mRNA expression profile. With this study, we aimed to demonstrate how LLDT-8 treatment significantly impacts the process of immune regulation. …
Continue reading at (5R)-5-Hydroxytriptolide (LLDT-8) induces substantial epigenetic mediated immune response network changes in fibroblast-like synoviocytes from rheumatoid arthritis patients
…
NCBIBuild 35 ==> hg17; NCBIBuild 36 ==> hg18; NCBIBuild 37 ==> hg19; NCBIBuild 38 ==> hg38
- All the figures are only used for non-profit education. reminding me if infrigement happens
How to generate dbSNP153(hg19) from dbSNP153(hg38,GRCH38)
Recently (07/09/2019), dbSNP have been updated dbSNP153 from dbSNP152. However, NCBI only provided dbSNP153 in hg38 (GRCH38) version without any source for hg19 version. Here, I prepared a approach to generate dbSNP153 in hg19..
dbSNP153 in hg19, GRCH37
wget https://ftp.ncbi.nih.gov/snp/redesign/latest_release/VCF/GCF_000001405.25.gz
wget https://ftp.ncbi.nih.gov/snp/redesign/latest_release/VCF/GCF_000001405.25.gz.tbi
wget https://raw.githubusercontent.com/Shicheng-Guo/AnnotationDatabase/master/GCF_000001405.25_GRCh37.p13_assembly_report.txt
awk -v RS="(\r)?\n" 'BEGIN { FS="\t" } !/^#/ { if ($10 != "na") print $7,$10; else print $7,$5 }' GCF_000001405.25_GRCh37.p13_assembly_report.txt > dbSNP-to-UCSC-GRCh37.p13.map
perl -p -i -e '{s/chr//}' dbSNP-to-UCSC-GRCh37.p13.map
bcftools annotate --rename-chrs dbSNP-to-UCSC-GRCh37.p13.map GCF_000001405.25.gz -Oz -o dbSNP153.hg19.vcf.gz
bcftools view dbSNP153.hg19.vcf.gz -Ov -o dbSNP153.hg19.vcf
dbSNP153 in hg38, GRCH38
Bash, Perl, Python and (GATK or CrossMap), awk and wget are required in this approach. Crossmap is Python based method which I don’t recommend to use since the version problem may waste tons of your time. I prefer to use GATK4 LiftoverVcf.
wget https://ftp.ncbi.nih.gov/snp/redesign/latest_release/VCF/GCF_000001405.38.gz
wget https://ftp.ncbi.nih.gov/snp/redesign/latest_release/VCF/GCF_000001405.38.gz.tbi
wget https://raw.githubusercontent.com/Shicheng-Guo/AnnotationDatabase/master/GCF_000001405.38_GRCh38.p12_assembly_report.txt
awk -v RS="(\r)?\n" 'BEGIN { FS="\t" } !/^#/ { if ($10 != "na") print $7,$10; else print $7,$5 }' GCF_000001405.38_GRCh38.p12_assembly_report.txt > dbSNP-to-UCSC-GRCh38.p12.map
bcftools annotate --rename-chrs dbSNP-to-UCSC-GRCh38.p12.map GCF_000001405.38.gz | gawk '/^#/ && !/^##contig=/ { print } !/^#/ { if( $1!="na" ) print }' | bgzip -c > GCF_000001405.38.dbSNP153.GRCh38p12b.GATK.vcf.gz
gatk LiftoverVcf -I GCF_000001405.38.dbSNP152.GRCh38p12b.GATK.vcf.gz -O dbSNP153.hg19.vcf -C hg38ToHg19.over.chain.gz --REJECT rejected.vcf -R ~/hpc/db/hg19/hg19.fa
liftOver with python CrossMap.py
python CrossMap.py vcf hg38Tohg19.over.chain.gz GCF_000001405.38.dbSNP153.GRCh38p12b.GATK.vcf.gz ~/hpc/db/hg19/hg19.fa GCF_000001405.38.dbSNP153.hg19.gz
perl -p -i -e '{s/chr//}' dbSNP-to-UCSC-GRCh38.p12.map
02/16/2020
wget https://ftp.ncbi.nih.gov/snp/redesign/latest_release/VCF/GCF_000001405.25.gz
wget https://ftp.ncbi.nih.gov/snp/redesign/latest_release/VCF/GCF_000001405.25.gz.tbi
wget https://raw.githubusercontent.com/Shicheng-Guo/AnnotationDatabase/master/GCF_000001405.25_GRCh37.p13_assembly_report.txt
awk -v RS="(\r)?\n" 'BEGIN { FS="\t" } !/^#/ { if ($10 != "na") print $7,$10; else print $7,$5 }' GCF_000001405.25_GRCh37.p13_assembly_report.txt > dbSNP-to-UCSC-GRCh37.p13.map
perl -p -i -e '{s/chr//}' dbSNP-to-UCSC-GRCh37.p13.map
bcftools annotate --rename-chrs dbSNP-to-UCSC-GRCh37.p13.map GCF_000001405.25.gz | gawk '/^#/ && !/^##contig=/ { print } !/^#/ { if( $1!="na" ) print }' | bgzip -c > dbSNP153.hg19.vcf.gz
03/11/2020 (again, I moved to UW-Madison, department of Medical Genetics, I need to rebuild the dbSNP153 in deepThought server)
Compare plink bim to 1000G/HRC dataset
### Script to check plink .bim files against HRC/1000G for strand, id names, positions, alleles,
### ref/alt assignment, William Rayner 2015, wrayner@well.ox.ac.uk, Version 4.2.7
cd /mnt/sas0/AD/sguo234/asa
wget http://www.well.ox.ac.uk/~wrayner/tools/HRC-1000G-check-bim-v4.2.7.zip
wget http://ngs.sanger.ac.uk/production/hrc/HRC.r1-1/HRC.r1-1.GRCh37.wgs.mac5.sites.tab.gz
wget https://www.well.ox.ac.uk/~wrayner/tools/1000GP_Phase3_combined.legend.gz
gunzip 1000GP_Phase3_combined.legend.gz
unzip HRC-1000G-check-bim-v4.2.7.zip
gunzip HRC.r1-1.GRCh37.wgs.mac5.sites.tab.gz
wget http://qbrc.swmed.edu/zhanxw/software/checkVCF/checkVCF-20140116.tar.gz
tar xzvf checkVCF-20140116.tar.gz
## compare bim to HRC European Population(HEP)
perl HRC-1000G-check-bim.pl -b RA3000.R3.bim -f RA3000.R3.frq -r HRC.r1-1.GRCh37.wgs.mac5.sites.tab -h
sh Run-plink.sh
## compare bim to 1000G East-Asian Population(EAP)
perl HRC-1000G-check-bim.pl -b RA3000.R3.bim -f RA3000.R3.frq -r 1000GP_Phase3_combined.legend -g -p EAS
sh Run-plink.sh
addfakegenotype to dbSNP153 and then transfer to plink format
wget https://raw.githubusercontent.com/Shicheng-Guo/Gscutility/master/addfakegenotype.pl
perl addfakegenotype.pl > dbSNP153.hg19.plink.vcf
plink --vcf dbSNP153.hg19.plink.vcf --make-bed --allow-extra-chr --out dbSNP153.hg19
How to share dbSNP153(hg19,GRCH37) and how to download dbSNP153(hg19,GRCH37)
In order to share dbSNP153(hg19,GRCH37), I uploaded dbSNP153(hg19,GRCH37) to . You can download dbSNP153(hg19,GRCH37) with the follow link. Good luck for your analysis. Finally, Thanks to Dr. Raony Guimarães for the help on the ideas of CrossMap to liftover vcf files..
Recessive Diplotype Identify Novel Hemochromatosis Gene
Recently, our project collaborated with Dr. Jiucun Wang named A gene-based recessive diplotype exome scan discovers FGF6, a novel hepcidin-regulating iron-metabolism gene was accepted by Blood
Standard analyses applied to genome-wide association data are well designed to detect additive effects of moderate strength. However, the power for standard genome-wide association study (GWAS) analyses to identify effects from recessive diplotypes is not typically high. We proposed and conducted a gene-based compound heterozygosity test to reveal additional genes underlying complex diseases. With this approach applied to iron overload, a strong association signal was identified between the fibroblast growth factor–encoding gene, FGF6, and hemochromatosis in the central Wisconsin population. Functional validation showed that fibroblast growth factor 6 protein (FGF-6) regulates iron homeostasis and induces transcriptional regulation of hepcidin. Moreover, specific identified FGF6 variants differentially impact iron metabolism. In addition, FGF6 downregulation correlated with iron-metabolism dysfunction in systemic sclerosis and cancer cells. Using the recessive diplotype approach revealed a novel susceptibility hemochromatosis gene and has extended our understanding of the mechanisms involved in iron metabolism. …
Authors of the study are Shicheng Guo#, Shuai Jiang#, Narendranath Epperla, Yanyun Ma, Mehdi Maadooliat, Zhan Ye, Brent Olson, Minghua Wang, Terrie Kitchner, Jeffrey Joyce, Peng An, Robert Strenn, Joseph J. Mazza, Fudi Wang, Jennifer K. Meece, Wenyu Wu, Li Jin, Judith A. Smith, Jiucun Wang* and Steven J. Schrodi*. This study was supported by the Clinical and Translational Science Award (CTSA) program (1UL1RR025011), the National Center for Advancing Translational Sciences (NCATS) grant (9U54TR000021), NCATS grant UL1TR000427 and Marshfield Clinic Research Institute grant SCH10218 and generous donors to the Marshfield Clinic Health System. Authors contributed equally to the work, *Corresponding authors: Dr. Jiucun Wang and Dr. Steven J. Schrodi. …
Some other papers need to be read related to this project:
- Properties and Modeling of GWAS when Complex Disease Risk Is Due to Non-Complementing, Deleterious Mutations in Genes of Large Effect
Continue reading at https://www.nature.com/articles/s41598-019-47411-1

- All the figures are only used for non-profit education. reminding me if infrigement happens
Next generation protocol to bcftools in medical genetics research
Today, I will give a talk about “Next generation protocol to bcftools in medical genetics research” in MCRI research hub meeting. As we know, bcftools, vcftools, plink2, GATK4 have been widely used in medical genetics and population genetics research. The usage of these tools require lots of experiences. However, the original protocols are quite limited espeically lacking of real-data example. Here, I will provide the real-data examples and solution to most frequently problem we meet in the usage of these tools.
bcftools view
bcftools view is the most frequent command to use for SNPs filtering, sample filtering, format changing.
bcftools view -i '(IMP=1 & R2>0.6)|IMPUTED=0' chr$i.dose.dbSNP.hg19.vcf.gz | bcftools annotate -x ^FORMAT/GT -Oz -o chr$i.dose.dbSNP.clean.hg19.vcf.gz
bcftools view -i \'R2\>0.6\|TYPED=1\|TYPED_ONLY=1\' -Oz chr$i.dose.vcf.gz -Oz -o chr$i.dose.filter.vcf.gz
bcftools annotation
# collect gene regions. don't forget to extend regions with -5K to +5K to region regions to cover promoter and enhancer SNPs
awk '{print $1,$2-5000,$3+5000,$4}' OFS="\t" MUC.hg19.bed | bedtools sort -i > MUC.hg19.sort.bed
bcftools annotate -a ~/hpc/db/hg19/dbSNP152/dbSNP152.chr$i.hg19.vcf.gz -c ID chr$i.dose.contig.vcf.gz -Oz -o chr$i.dose.dbSNP.hg19.vcf.gz >>$i.job
# merge MUC genotypes from chr1 to chr22
cd /gpfs/home/guosa/hpc/rheumatology/RA/he2020/impute/R3
ls chr*.dose.MUC.clean.hg19.vcf.gz > MUC.vcf.txt
bcftools concat -f MUC.vcf.txt -Oz -o MUC.hg19.vcf.gz
bcftools annotate -a ~/hpc/db/hg19/dbSNP152/dbSNP152.chr$i.hg19.vcf.gz -c ID chr$i.dose.contig.vcf.gz -Oz -o MUC.hg19.vcf.gz
# https://github.com/Shicheng-Guo/AnnotationDatabase/blob/master/hg19/refGene.hg19.VCF.sort.bed.gz
# https://github.com/Shicheng-Guo/AnnotationDatabase/blob/master/hg19/refGene.hg19.VCF.sort.bed.gz.tbi
bcftools annotate -a ~/hpc/db/hg19/refGene.hg19.VCF.sort.bed.gz -c CHROM,FROM,TO,GENE -h <(echo '##INFO=<ID=GENE,Number=1,Type=String,Description="Gene name">') MUC.hg19.vcf.gz -Oz -o MUC.anno.hg19.vcf.gz
# review dbSNPs from vcf.gz file
bcftools view -i '%iD=="rs35705950"' MUC.anno.hg19.vcf.gz | less -S
bcftools view -i '%iD=="rs79920422"' MUC.anno.hg19.vcf.gz | less -S
How to fix this strand flips for Michigan Imputation Server?
- Error: More than 100 obvious strand flips have been detected. Please check strand. Imputation cannot be started! ``` chr2.1kg.phase3.v5a.dedup.norm.EUR.vcf.gz zcat chr2.1kg.phase3.v5a.dedup.norm.EUR.vcf.gz | awk ‘{print $1,$2,$3,$4,$5}’ OFS=”\t”| grep -v ‘.;’ > keep.txt bcftools view -T keep.txt chr2.1kg.phase3.v5a.dedup.norm.EUR.vcf.gz -Oz -o chr2.1kg.phase3.v5a.dedup.norm.clean.EUR.vcf.gz
you need remove the following SNPs from chr2.1kg.phase3.v5a.dedup.EUR.vcf.gz
^2 18004148 .;rs541466601 AGAGCCCA AGAGCCCG ^2 33172927 .;rs554173463 GC GA ^2 40484170 .;rs537996337 AAATAAATA AAATAAATG ^2 44234879 rs550419970;.;rs533534296 ATAAA ATAAATAAA ^2 45515468 .;rs544467622 CTTTTGT CTTTTAT ^2 47982412 .;rs536566627 GG GC ^2 74559847 .;rs564494041 TGT TGC ^2 77195037 .;rs548983836 TTAC TCAC ^2 84495638 rs573607330;.;rs569169043 AATAA AATAAATAA ^2 109192726 .;rs539410502 GTTTTGTTTTG GTTTTGTTTTC ^2 179073443 .;rs538697958 TTTTCC TTTGCC ^2 206417755 rs574286642;.;rs576039119 AGAA AGAAGAA
### vcftools
zcat dbSNP152.chr1.hg19.vcf.gz | vcf-sort -p 16 -t ./temp/ | bgzip -c > dbSNP152.chr1.hg19.sort.vcf.gz & zcat dbSNP152.chr7.hg19.vcf.gz | vcf-sort -p 16 -t ./temp/ | bgzip -c > dbSNP152.chr7.hg19.sort.vcf.gz & zcat dbSNP152.chr8.hg19.vcf.gz | vcf-sort -p 16 -t ./temp/ | bgzip -c > dbSNP152.chr8.hg19.sort.vcf.gz & zcat dbSNP152.chr9.hg19.vcf.gz | vcf-sort -p 16 -t ./temp/ | bgzip -c > dbSNP152.chr9.hg19.sort.vcf.gz &
#### GATK
Some of my colleagues meet lots of GATK bugs. Please be sure GATK requires `Java 1.8` other Java will have some unexpected errors. On the other side, please download the database from [GATK Resource Bundle ftp server](https://software.broadinstitute.org/gatk/download/bundle) rather than other database.
gatk CreateSequenceDictionary -R hg19.fa -O hg19.dict
#### SnpSift
#### Example 1. How to build vcf annotation database for bcftools annotate
hg19 dbSNP
wget ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh37p13/VCF/All_20180423.vcf.gz wget ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh37p13/VCF/All_20180423.vcf.gz.md5 wget ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh37p13/VCF/All_20180423.vcf.gz.tbi
hg38 dbSNP
wget ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh38p7/VCF/All_20180418.vcf.gz wget ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh38p7/VCF/All_20180418.vcf.gz.md5 wget ftp://ftp.ncbi.nih.gov/snp/organisms/human_9606_b151_GRCh38p7/VCF/All_20180418.vcf.gz.tbi
copy files from UW-Madison to MCRI
scp nu_guos@submit-1.chtc.wisc.edu:/home/nu_guos/All_20180423* ~/hpc/db/hg19/dbSNP
split with chrosome name
SNP orders are not correct, sort chr1,chr7,chr8,chr9
mkdir ./temp/chr1/ mkdir ./temp/chr7/ mkdir ./temp/chr8/ mkdir ./temp/chr9/ zcat dbSNP152.chr1.hg19.vcf.gz | vcf-sort -p 16 -t ./temp/ | bgzip -c > dbSNP152.chr1.hg19.sort.vcf.gz & zcat dbSNP152.chr7.hg19.vcf.gz | vcf-sort -p 16 -t ./temp/ | bgzip -c > dbSNP152.chr7.hg19.sort.vcf.gz & zcat dbSNP152.chr8.hg19.vcf.gz | vcf-sort -p 16 -t ./temp/chr8 | bgzip -c > dbSNP152.chr8.hg19.sort.vcf.gz & zcat dbSNP152.chr9.hg19.vcf.gz | vcf-sort -p 16 -t ./temp/chr9 | bgzip -c > dbSNP152.chr9.hg19.sort.vcf.gz & mv dbSNP152.chr8.hg19.sort.vcf.gz dbSNP152.chr8.hg19.sort.vcf.gz mv dbSNP152.chr9.hg19.sort.vcf.gz dbSNP152.chr9.hg19.sort.vcf.gz tabix -p vcf dbSNP152.chr8.hg19.sort.vcf.gz & tabix -p vcf dbSNP152.chr9.hg19.sort.vcf.gz & ```
Footnote:
-
human refGene hg19 TSS: https://raw.githubusercontent.com/Shicheng-Guo/AnnotationDatabase/master/hg19/refGene_hg19_TSS.bed
-
human refGene hg38 TSS: https://raw.githubusercontent.com/Shicheng-Guo/AnnotationDatabase/master/hg38/refGene.hg38.TSS.bed
-
All the figures are only used for non-profit education. reminding me if infrigement happens
How to prepare a computational biology work station in MCRI
One of terrible things in marshfield clinic research institute (MCRI) is that software/package install request. Any software even Rstudio, R, Python will be reviewed by ITS department for security issues. Furthermore, the review process usually takes 2-3 months which make the research become quite difficult. Lucky thing is we can submit request as soon as possible supposing you think the software will be potentially used in the coming month. Finally, ITS don’t provide the software list which have been approved. therefore, it is quite necessary to list them and I think it will be helpful for further research fellows in MCRI.
Here are software/packages I requested in the past two years and I will real-time update the list:
- R
- R-studio
- Python
- pycharm
- Perl
- BioEdit
- Eclipse+EPIC
- Endnote
- Adobe Illustrator/inkscape
- Adobe Photoshop
- MEGA7
- Filezilla
- Cytoscape
- Haploview
- Notepad++
- NCBI-Cn3D
- MiKTex
- WINSCP
- Mendeley
- Evernote/Zotero
- Zoom
- IGV/IGVtools
- Julia
- Rust
- GATK
- Picard
- Deep Variant
-
Dragen
- All the figures are only used for non-profit education. reminding me if infrigement happens
How to apply deeptools for Medip-seq and MBD-seq analysis
deepTools is a suite of python tools particularly developed for the efficient analysis of high-throughput sequencing data, such as ChIP-seq, RNA-seq or MNase-seq. deeptools has been widely applied in bam/bigwig data analysis. here, I show some example how to use deeptools in mbd-seq and medip-seq methylation data analysis. Meanwhile, MACS usage will also be shown in this poster.
Genome-wide multiple bedgraph data analysis with Intervene
Today, I will give a talk about how to do multiple bedgraph data analysis with Intervene for ChIP-seq or MBD-seq data with Intervene. Intervene is a tool for intersection and visualization of multiple genomic region and gene sets (or lists of items). Intervene provides an easy and automated interface for effective intersection and visualization of genomic region sets or lists of items, thus facilitating their analysis and interpretations.
conda install -c bioconda intervene
for i in 2019032901 2019032903 2019040901 2019051703 2019052301 2019053101 2019053102
do
intervene venn -i /gpfs/home/guosa/hpc/methylation/pancrease/medip/venn/$i*.bed --project $i
done
Here, you can find the venn diagrams which are quite fancy. We can prepare venn for each sample.