Shicheng Guo Genetic and Epigenetic Research to Human Complex Diseases

-
23 Jan 2020 » Novel 2019 coronavirus genome in Wuhan, China and USA
- Janssen Vaccine start phase 1/2 clinical trials……..
- SARS-CoV (accession no. NC_004718): https://www.ncbi.nlm.nih.gov/nuccore/NC_004718.3?report=fasta
- Virus variants reflect patients severity: https://www.medrxiv.org/content/10.1101/2020.04.14.20060160v1.full.pdf+html
- 04/13/2020: xxx whole genome of COV19 were downloaded and you can download here
- 04/12/2020: Effective reproductive number (R) decrease from 3.0 to 0.3 with cordons sanitaire, traffic restriction, social distancing, home confinement, centralized quarantine, and universal symptom survey.
- 04/05/2020: The COVID-19 host genetics initiative: https://covid19hg.netlify.com/
- 04/05/2020: SITC Statement on anti-IL-6/IL-6R for COVID-19, use of IL-6 or IL-6-receptor (IL-6R) blocking antibodies like tocilizumab (Actemra, Roche-Genentech), sarilumab (Kevzara, Regeneron) and siltuximab (Sylvant, EUSA Pharma)
- 04/01/2020: How The Body Reacts To Viruses from Harvard University video
- 03/30/2020: Susceptibility of ferrets, cats, dogs, and different domestic animals to SARS-coronavirus-2
- 03/30/2020: Janssen works together with Harvard using Janssen patented technique: AdVac and PER.C6 to develop COVID19 vaccines.
- 03/30/2020: Janssen and Biomedical Advanced Research and Development Authority (BARDA) for 1 billion $ Vaccines development
- 03/12/2020: ACE2 and TMPRSS2: The novel coronavirus 2019 (2019-nCoV) uses ACE2 and TMPRSS2 to entry target cells
- 03/06/2020:Covid-19 Small Molecule Therapies Reviewed from Science Translational Medicine
- 03/07/2020: FTF to download Virus Genome: ftp://download.big.ac.cn/Genome/Viruses/Coronaviridae/genome/
- 03/07/2020: Virus Genome Download Update: https://bigd.big.ac.cn/ncov/release_genome and https://www.gisaid.org/
- 03/03/2020: How is AI Informing the Global Health and Business Response to 2019-nCOV?
- 03/03/2020: Technical Problems with Existing CDC COVID-19 Primers, and an Improved Set of Primers.
- 02/03/2020: Dr. Zheng-Li Shi published A pneumonia outbreak associated with a new coronavirus of probable bat origin in Nature.
- 02/03/2020: Dr. Yong-Zhen Zhang lab in IBS,Fudan University published A new coronavirus associated with human respiratory disease in China in Nature
- 02/03/2020: demographics, clinical information and reason of infection were available for the first 2020 patents, click here to download
- 02/02/2020: 54 nCoV RNAs are available: https://raw.githubusercontent.com/Shicheng-Guo/2019nCoV/master/2019_nCov_genomes_2020.02.02.fasta
- 01/30/2020: Kaggle online: Daily level information on the number of 2019-nCoV affected cases across the globe:
- 01/25/2020: German Prof. Rolf Hilgenfeld, structure biologist, bring his coronavirus Substances of inhibitors to Wuhan to test the efficacy, see nature news.
- 01/24/2020: Host and infectivity prediction of Wuhan 2019 novel coronavirus using deep learning algorithm
- 01/23/2020: Discovery of a novel coronavirus associated with the recent pneumonia outbreak in 3 humans and its potential bat origin
- 01/19/2020: A mathematical model for simulating the transmission of Wuhan novel Coronavirus, see bioRxiv
- 01/24/2020: One way that viruses adapt is by encoding proteins using the same choice of codons as their host??
- 01/24/2020: primary host -> intermediate host -> human, secondary host require long time to accumulate mutation
- 01/24/2020: more discussion about 2019-nCoV in virological: http://virological.org/t/novel-2019-coronavirus-genome/319
- 01/24/2020: one-off Nextstrain build for SARS-like coronaviruses from Bedford Lab: https://github.com/blab/sars-like-cov
- 01/24/2020: ACE2 and TMPRSS2: Angiotensin I Converting Enzyme 2, functional receptor for the spike glycoprotein of the human coronaviruses SARS and HCoV-NL63.
- 01/23/2020: Phylogeny of 6 SARS-like betacoronaviruses in Wuhan.
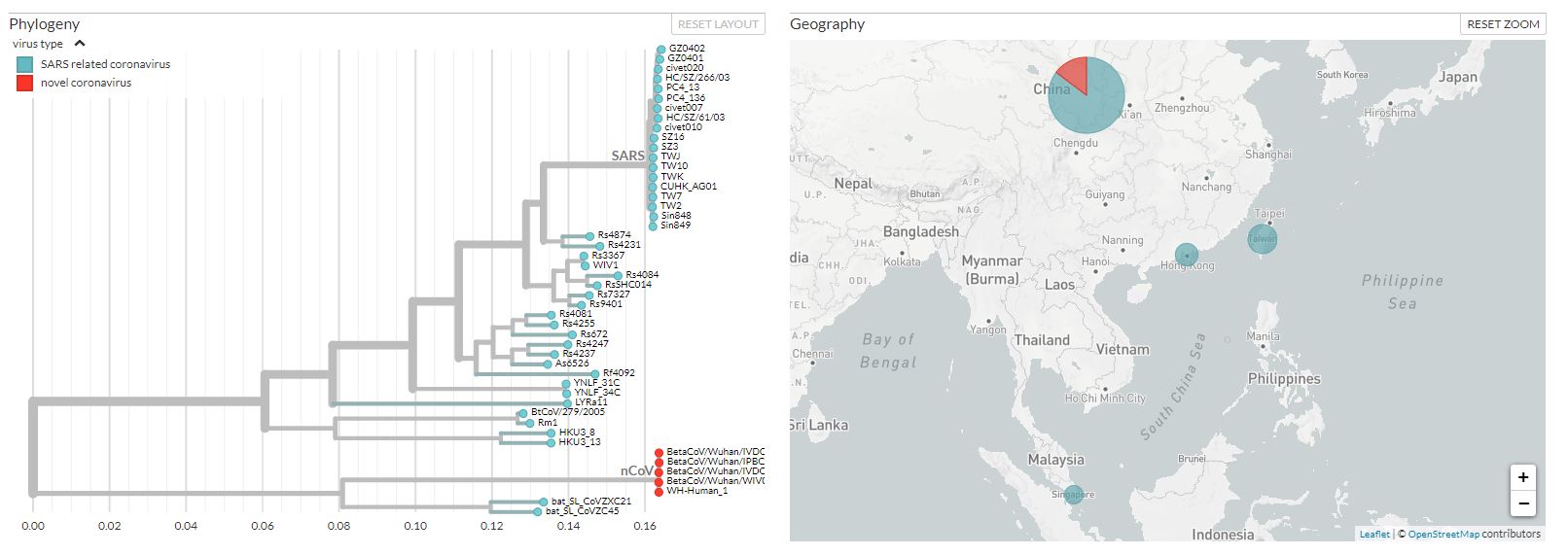
- 01/23/2020: Five new genomes have been deposited in the GISAID platform: https://gisaid.org/CoV2020
- 01/23/2020: USA confirmed the first 2019-nCoV occurred in USA in the state of Washington
- 01/23/2020: Upload fasta file to 2019nCoV and compared with other nCoV
- 01/22/2020: Dr. Shi identified bat as the virus host and upload the paper to bioRxiv
- 01/10/2020: Fudan University shared the sequence to GenBank with accession MN908947
- 12/12/2019: the first 2019-nCoV occurred in Wuhan, China and scientist think the earliest time might be 11/20/2019
-
20 Jan 2020 » Robust Region-Based Rare-Variant Test to UKBB-Seq Data
-
08 Nov 2019 » Genome-wide DNA methylation to human atrial fibrillation
-
30 Oct 2019 » Genome-wide bisulfite sequencing to human tissue samples
-
28 Oct 2019 » Epigenome Research to Human Cancers with WGBS
-
28 Oct 2019 » Genomics, Environment Interaction, Epigenetics and Aging
- McEWen, PNAS, 2019, The PedBE clock accurately estimates DNA methylation age in pediatric buccal cells
- Stubbs, Genome Biol, 2017,Multi-tissue DNA methylation age predictor in mouse.
- Horvath, Genome Biol, 2013, DNA methylation age of human tissues and cell types.
-
26 Oct 2019 » Population Genetics in East Asian and Allele Frequency
-
24 Oct 2019 » Genetic and Epigenetic based Intra-Tumor Heterogeneity
-
12 Oct 2019 » Electronic health/medical records (EHR/EMR) data analysis
-
08 Oct 2019 » US-FDA Artificial Intelligence and Machine Learning Discussion
-
07 Oct 2019 » Artificial intelligence in risk prediction and diseases diagnosis
-
01 Oct 2019 » How to design and conduct a biomedical research in MCRI
-
19 Aug 2019 » Genome-wide cell-free DNA fragmentation in patients with cancer
-
01 Aug 2019 » (5R)-5-Hydroxytriptolide and Epigenetics of Rheumatoid Arthritis
-
09 Jul 2019 » How to generate dbSNP153(hg19) from dbSNP153(hg38,GRCH38)
-
28 Feb 2019 » Recessive Diplotype Identify Novel Hemochromatosis Gene
-
01 Oct 2018 » Next generation protocol to bcftools in medical genetics research
-
27 Feb 2018 » How to prepare a computational biology work station in MCRI
-
11 Oct 2017 » How to apply deeptools for Medip-seq and MBD-seq analysis
-
30 Sep 2017 » Genome-wide multiple bedgraph data analysis with Intervene
-
01 Aug 2017 » How to update hapmap2 and hapmap3 from hg18 to hg19 or hg38
-
28 Jun 2017 » Characterizing population with principle componment analysis
-
13 Jun 2017 » Data Science in Population Genetics and Medical Genetics
-
10 Jun 2017 » Geneics and epigeneitcs of rheumatoid arthritis and osteoarthritis
-
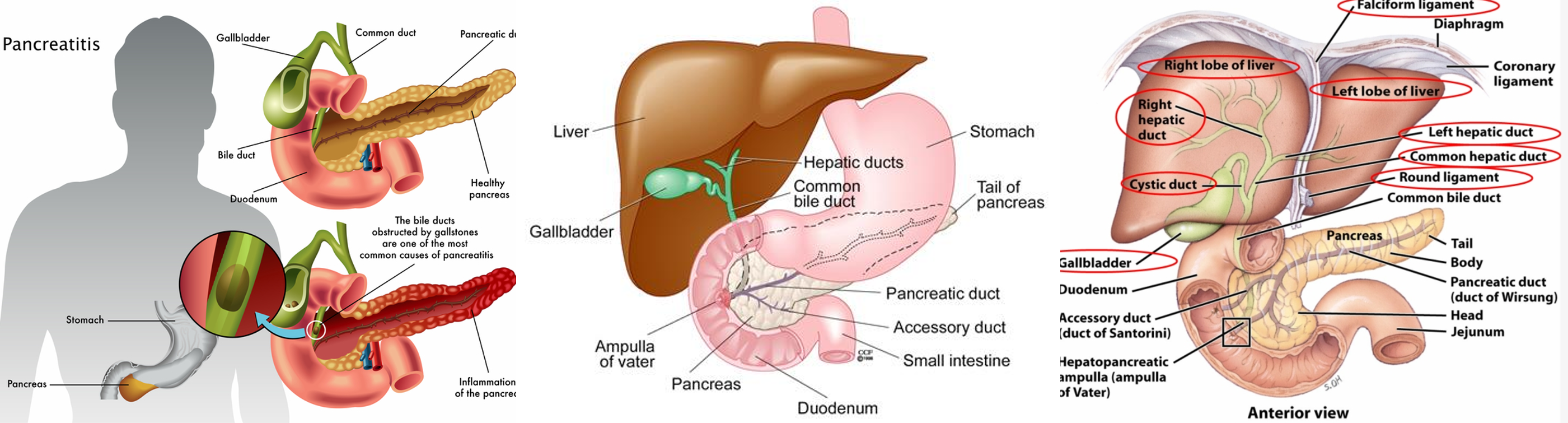
09 Jun 2017 » Genetics and epigenetics of pancreatic and cholangiocarcinoma
- Pancreatic cancer and cholangiocarcinoma
- All the figures are only used for non-profit education. reminding me if infrigement happens.
-
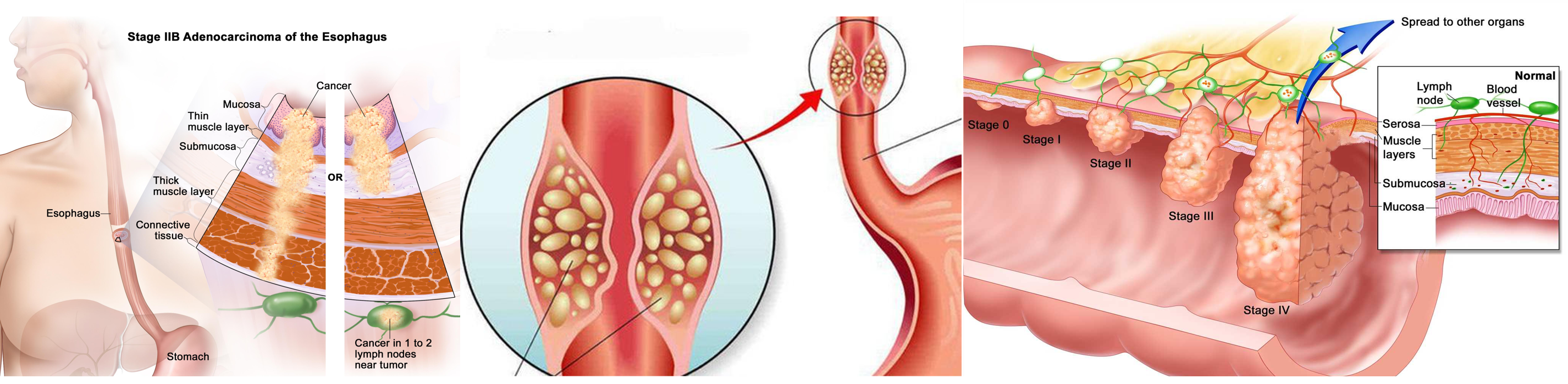
28 May 2017 » Genetic and epigenetic of esophageal squamous cell carcinoma
- Esophageal squamous cell carcinoma
- All the figures are only used for non-profit education. reminding me if infrigement happens.
-
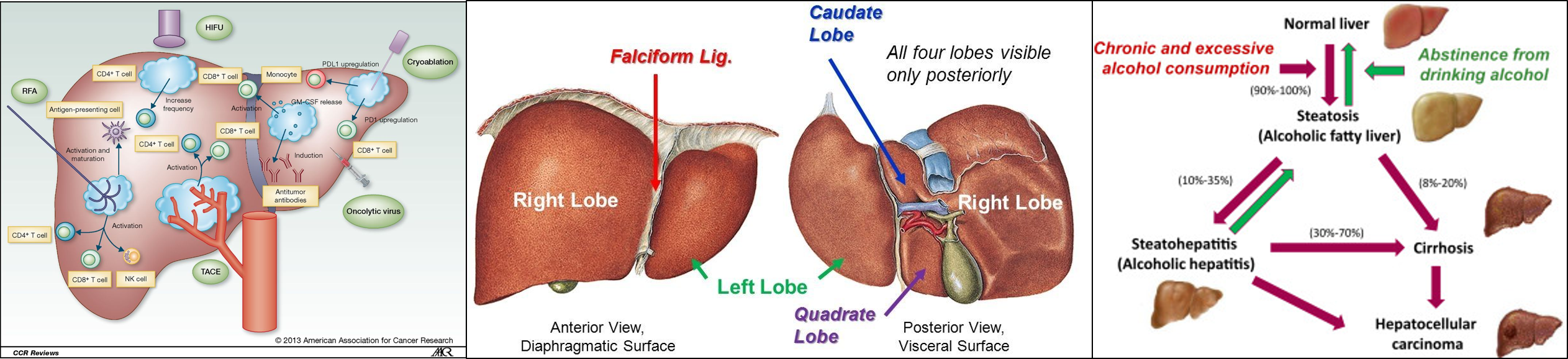
02 May 2017 » Advances in Genetic and epigenetic of Hepatocellular Carcinoma
- Advances in Genetic and epigenetic of Hepatocellular Carcinoma
- All the figures are only used for non-profit education. reminding me if infrigement happens
-
01 May 2017 » How to apply R to prepare fancy figures in the manuscript
- All the figures are only used for non-profit education. reminding me if infrigement happens.
-
23 Apr 2017 » Explaining complex machine learning models with LIME
-
09 Apr 2017 » How to apply R for Plotting 3D maps and location tracks
-
03 Apr 2017 » Science Writing: Guidelines And Guidance in Medical Research
-
02 Apr 2017 » Dealing with unbalanced data in machine learning
-
31 Mar 2017 » Building meaningful machine learning models for disease prediction
-
16 Mar 2017 » Plotting trees from Random Forest models with ggraph
-
07 Mar 2017 » Hyper-parameter Tuning with Grid Search for Deep Learning
-
27 Feb 2017 » Building deep neural nets with h2o and rsparkling that predict arrhythmia of the heart
-
19 Feb 2017 » Predicting food preferences with sparklyr (machine learning)
-
12 Feb 2017 » Conditional ggplot2 geoms in functions (QTL plots)
-
06 Feb 2017 » Scratching the Surface of Gender Biases
-
30 Jan 2017 » New features in World Gender Statistics app
-
29 Jan 2017 » Exploring World Gender Statistics with Shiny
-
22 Jan 2017 » R vs Python - a One-on-One Comparison
-
15 Jan 2017 » Feature Selection in Machine Learning (Breast Cancer Datasets)
-
05 Jan 2017 » Gene homology Part 3 - Visualizing Gene Ontology of Conserved Genes
-
30 Dec 2016 » How to map your Google location history with R
-
22 Dec 2016 » Animating Plots of Beer Ingredients and Sin Taxes over Time
-
18 Dec 2016 » How to build a Shiny app for disease- & trait-associated locations of the human genome
-
14 Dec 2016 » Gene homology Part 2 - creating directed networks with igraph
-
11 Dec 2016 » Creating a network of human gene homology with R and D3
-
04 Dec 2016 » How to set up your own R blog with Github pages and Jekyll Bootstrap
-
02 Dec 2016 » Extreme Gradient Boosting and Preprocessing in Machine Learning - Addendum to predicting flu outcome with R
-
27 Nov 2016 » Can we predict flu deaths with Machine Learning and R?
-
20 Nov 2016 » Analysing the Gilmore Girls' coffee addiction with R
-
13 Nov 2016 » Creating a Gilmore Girls character network with R
-
06 Nov 2016 » Is 'Yeah' Josh and Chuck's favorite word?
-
01 Nov 2016 » Exploring the human genome (Part 2) - Transcripts
-
23 Oct 2016 » Exploring the human genome - Gene Annotations
-
16 Oct 2016 » USA/ Canada Roadtrip 2016
-
29 Sep 2016 » DESeq2 Course Work
-
28 Sep 2016 » exprAnalysis package
-
08 Jan 1945 » Gene ID Transfer with R between Symbol, Gene ID and KEGG ID
- Transfer between Gene ID and Gene Symbol:
-
08 Jan 1944 » Human Population Genetics and related data operation
- How to download dbSNP153 in hg38 human genome reference
wget https://ftp.ncbi.nih.gov/snp/redesign/latest_release/VCF/GCF_000001405.38.gz wget https://ftp.ncbi.nih.gov/snp/redesign/latest_release/VCF/GCF_000001405.38.gz.md5 wget https://ftp.ncbi.nih.gov/snp/redesign/latest_release/VCF/GCF_000001405.38.gz.tbi wget https://ftp.ncbi.nih.gov/snp/redesign/latest_release/VCF/GCF_000001405.38.gz.tbi.md5 wget https://raw.githubusercontent.com/Shicheng-Guo/AnnotationDatabase/master/GCF_000001405.38_GRCh38.p12_assembly_report.txt gawk -v RS="(\r)?\n" 'BEGIN { FS="\t" } !/^#/ { if ($10 != "na") print $7,$10; else print $7,$5 }' GCF_000001405.38_GRCh38.p12_assembly_report.txt > dbSNP-to-UCSC-GRCh38.p12.map perl -p -i -e '{s/chr//}' dbSNP-to-UCSC-GRCh38.p12.map bcftools annotate --rename-chrs dbSNP-to-UCSC-GRCh38.p12.map GCF_000001405.38.gz | gawk '/^#/ && !/^##contig=/ { print } !/^#/ { if( $1!="na" ) print }' | bgzip -c > GCF_000001405.38.dbSNP153.GRCh38p12b.GATK.vcf.gz -
08 Jan 1943 » How to set up github and use it to accelerate your research
- if you are the first time to use github, what you need to do first are:
- Get a github account
- Download and install git.
- Set up git with your user name and email
- Now I am assume you use Linux/Ubuntu, I don’t use MAC
- Open a terminal/shell and type to config your github (name, email and color):
git config --global user.name "Shicheng-Guo" git config --global user.email "Shicheng.Guo@hotmail.com" git config --global color.ui true - you can make more config if you want, for example, if you want to use emacs:
git config --global core.editor emacs - the guide to setting up password-less logins (SSH keys connection).
ssh-keygen -t rsa -C "Shicheng.Guo@hotmail.com" less ~/.ssh/id_rsa.pub - Now you will see ssh key and copy it and paste to SSH Keys in your github Account Settings and test the setting with the following command. You can have multiple key for different computers so that all of them can be connected with your github account.
ssh -T git@github.com - Hope you can receive the following response
Hi username! You've successfully authenticated, but Github does not provide shell access.
- Open a terminal/shell and type to config your github (name, email and color):
- Okay, Now we start for the second step, to create new project or upload codes to existed project
-
08 Jan 1942 » Environment Setting in All My Previous Working Station
-
08 Jan 1941 » How to Install HTseq in Linux and STAR for RNA-seq
-
08 Jan 1940 » How to apply MACS2 to identify methylation peaks with MBD-seq
-
08 Jan 1939 » DNA methylation and Epigenetic research in Human Population
-
28 Feb 1938 » Multi-Omics of Characterization of the Cancer Cell Line Encyclopedia
- The CancerCell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity
- 01/22/2020: Quantitative Proteomics of the Cancer Cell Line Encyclopedia: https://gygi.med.harvard.edu/publications/ccle
- 01/22/2020: NCI60 proteome resource is a web application that facilitates comprehensive
- 05/23/2019: Next-generation characterization of the Cancer Cell Line Encyclopedia: https://doi.org/10.1038/s41586-019-1186-3
- 03/29/2012: The CancerCell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity
-
28 Feb 1937 » Dac Demethylation
-
28 Feb 1936 » Riblast_ncrna_target
-
28 Feb 1934 » Most Important Database for Human Biomedical Research
- Cancer Dependency Map, 2020, Identifying all dependencies in every cancer cell, https://depmap.org/portal/
- Open Targets is an innovative, large-scale, multi-year, public-private partnership that uses human genetics and genomics data for systematic drug target identification and prioritisation.
- VnD: a structure-centric database of disease-related SNPs and drugs: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3013797/
- Drugbank-databases: http://www.drugbank.ca/w/databases
- PharmGKB: https://www.pharmgkb.org/
-
28 Feb 1933 » How to get genomic coordinates for all protein domains
-
28 Feb 1931 » How to Understand Human Cancer
-
28 Feb 1930 » How to Submit NGS Seqeuencing data to SRA
-
28 Feb 1929 » Resources for flow cytometry bioinformatics analysis
-
28 Feb 1928 » How to use plink1.9/plink2 to do human genetics research
-
28 Feb 1928 » RNA-seq data to reveal novel response mechanism to bacterial
-
28 Feb 1928 » RNA-seq data with metaphlan2 to reveal novel response mechanism to bacterial
- metaphlan2 is based on python 2.7 and don’t require to download NR database since it use its own annotated database. if it is first time to comply py2, maybe you need restart the ternimal before install metaphlan2
-
28 Feb 1926 » EAS and SAS data in GenomeAsia100K panel
- STEP1: download GenomeAsia100K panel data
# GenomeAsia100K data are available at the links below. Users can download GA100K data in compressed Variant Call Format (VCF) file. mkdir ~/hpc/db/GenomeAsia100K cd ~/hpc/db/GenomeAsia100K for i in {1..22} do wget --no-check-certificate https://browser.genomeasia100k.org/service/web/download_files/$i.substitutions.annot.cont_withmaf.vcf.gz & done -
28 Feb 1925 » Genomics, Genomics and Related Disciplines
- Comparative genomics
- Functional genomics
- Population genomics
- Structural genomics
- Epigenomics
- Metagenomics
- Genomic medicine
- Synthetic biology and bioengineering
- Computational genomics
- Pathogenomics
- Personal genomics
- Transcriptomics
- Psychogenomics
-
28 Feb 1924 » How to reformat GeneSky GSA report to Plink
-
28 Feb 1923 » How to merge 7000 VCF files with bcftools merge?
-
28 Feb 1922 » Automatic GWAS and Post-GWAS Analysis Pipeline
-
28 Feb 1921 » How to install ANNOVAR in DeepThought@UW-Madison
-
28 Feb 1920 » How to Prepare Annotation DB Folder for Bioinformatics Analysis
-
28 Feb 1919 » How to Prepare China and USA Map with R
-
28 Feb 1918 » FDA and drug development pipeline
- J&J defends Darzalex with FDA approval for faster dosing
- Rcpi: R/Bioconductor Package as an Integrated Informatics Platform for Drug Discovery: https://www.bioconductor.org/packages/release/bioc/vignettes/Rcpi/inst/doc/Rcpi.html
- TAGRISSO: Targeted therapies to cancer that has tested positive for certain types of EGFR mutations. TAGRISSO specifically targets and blocks mutated EGFR found on cancer cells.
- Fragment-based lead discovery (FBLD) also known as fragment-based drug discovery (FBDD)
- BRAF is a human gene that encodes a protein called B-Raf. The gene is also referred to as proto-oncogene
- The Drug Development Process: The Drug Development Process
- In order to incentivize the development of new drugs, the FDA grants drug developers market exclusivity for a period of five years after a new drug is approved, during which time, no other firm is permitted to sell the drug, whether or not it is protected by a patent.
- China National PGx Data Science Consortium (CNPGx)
- KEGG DRUG: Methotrexate https://www.genome.jp/dbget-bin/www_bget?dr:D00142
-
28 Feb 1917 » How to Install Ensembl Variant Effect Predictor
- CRAN Task View: Meta-Analysis: https://cran.rstudio.com/web/views/MetaAnalysis.html
-
28 Feb 1916 » FDA approved drugs and the mechanism
- 2013: Bluebird Bio: autologous CD34+ hematopoietic stem cells transduced with LentiGlobin BB305 lentiviral vector encoding the human BA-T87Q-globin gene
-
28 Feb 1915 » Bioinformatics Skills Required
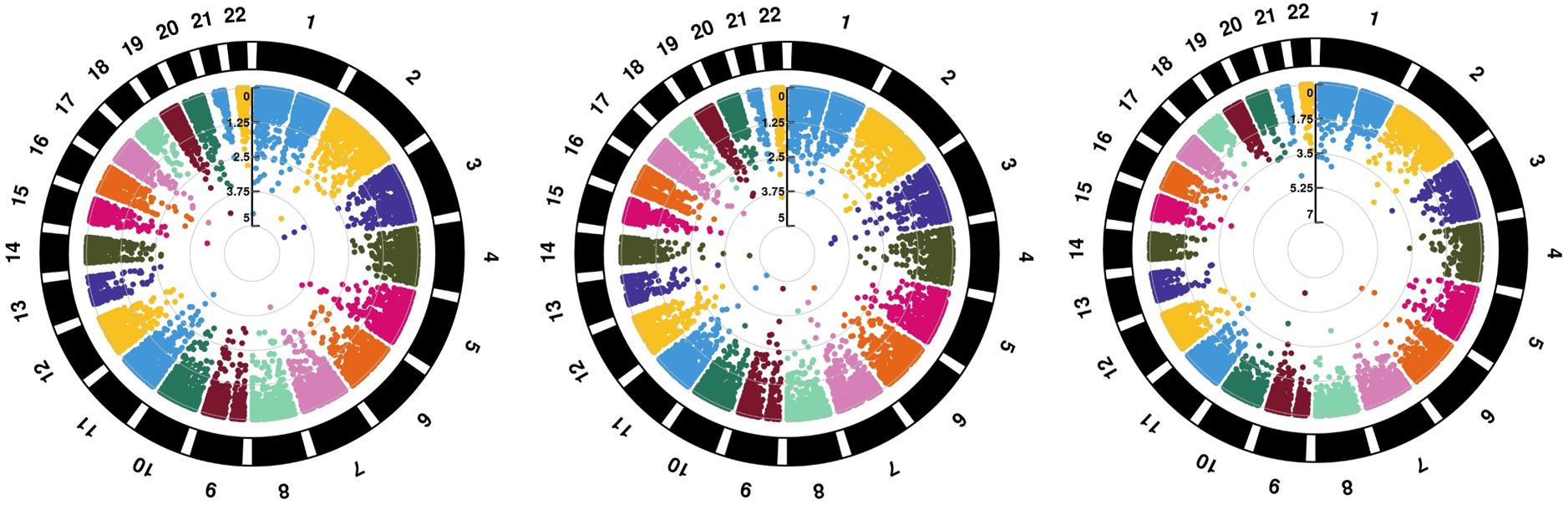
On 01/19/2020, I will give a talk about “UK Biobank Whole-Exome Sequence Binary Phenome Analysis with Robust Region-Based Rare-Variant Test, The American Journal of Human Genetics, December 19, 2019” in Marshfield Clinic Research Institute. Please check the PPT I prepared. I have implemented the pipeline in Marshfield Clinic SuperServer HPC cluster. The Figure I prepare for this post is GWAS result for colon cancer (CRC), RA and ESCC based on UKBB-50K-Exome-sequencing data. Several genes looks very interesting, for example SCL21A2.
Continue reading...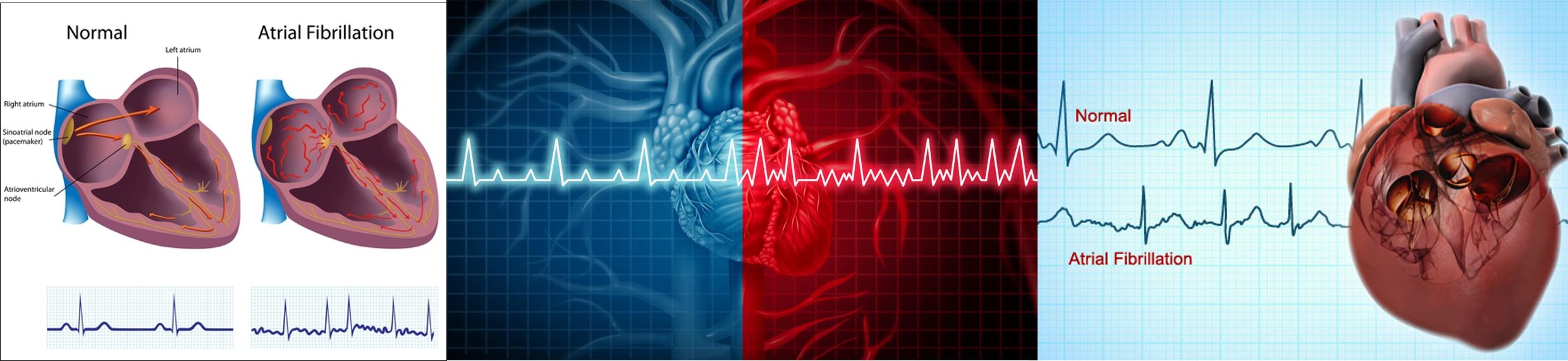
Aim and Background
Continue reading...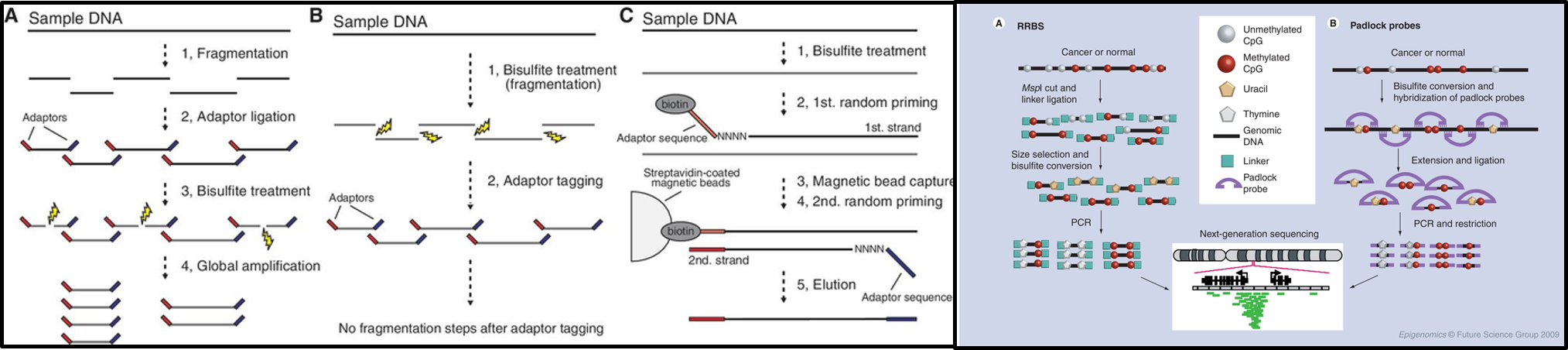
Aim and Background
Continue reading...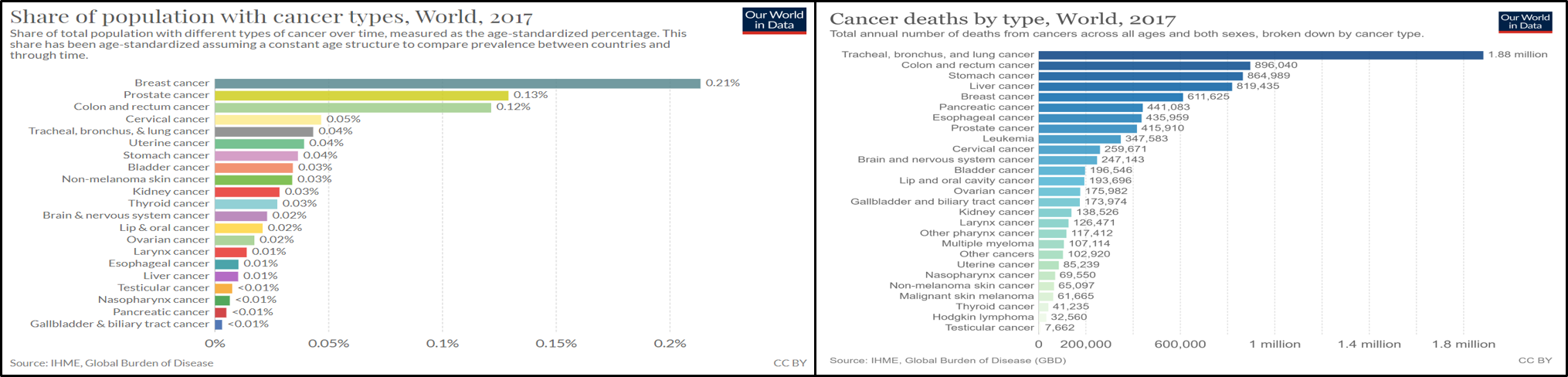
Here, I want to summarize Epigenome Research to Human Cancers with WGBS.
Continue reading...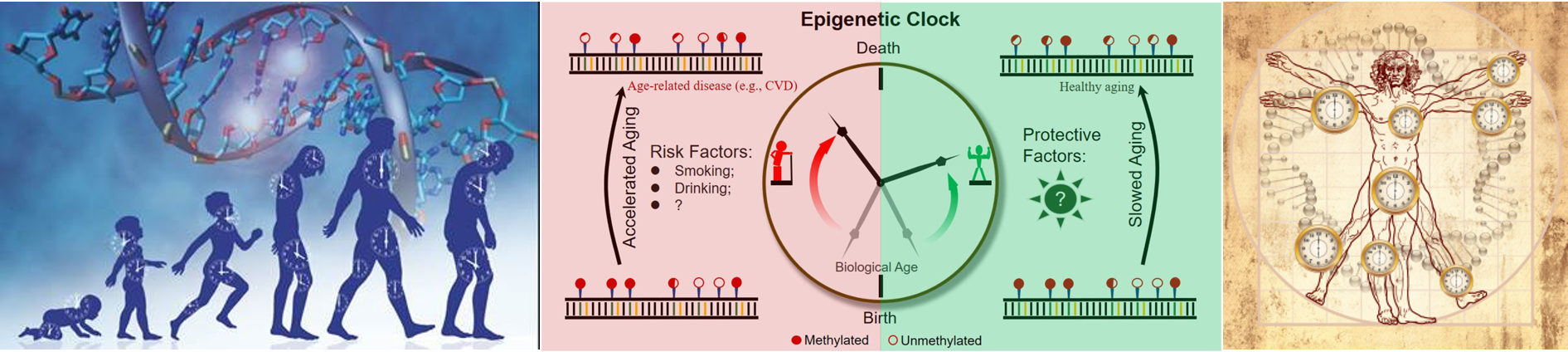
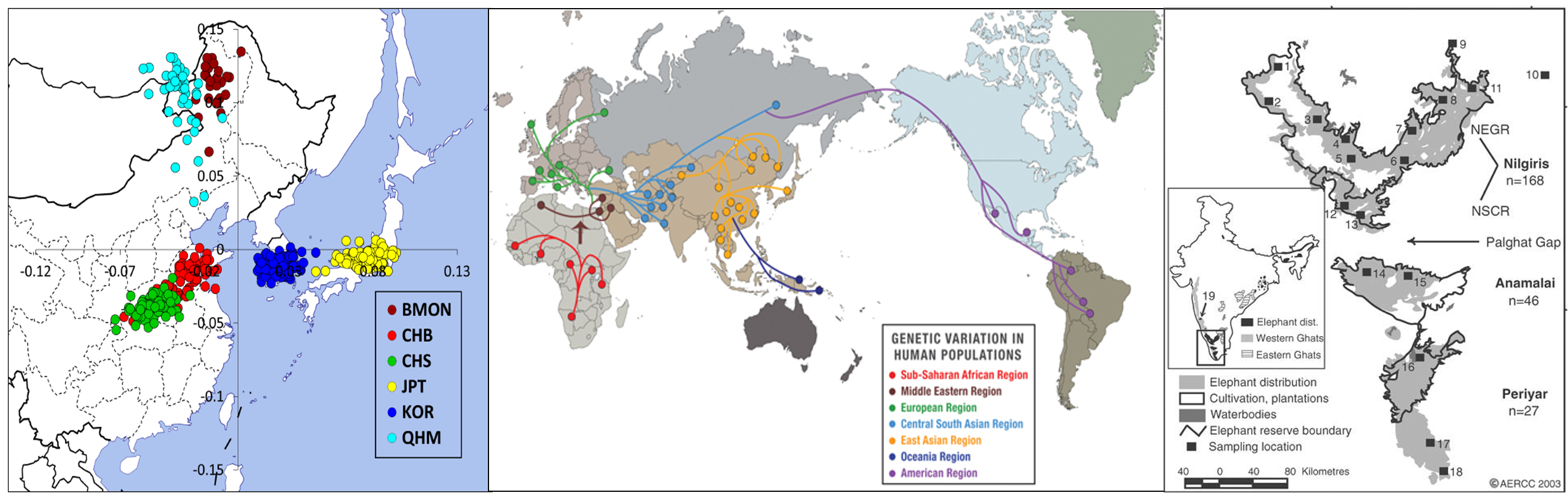
Here, I want to summarize Population Genetics in East Asian and Allele Frequency.
Continue reading...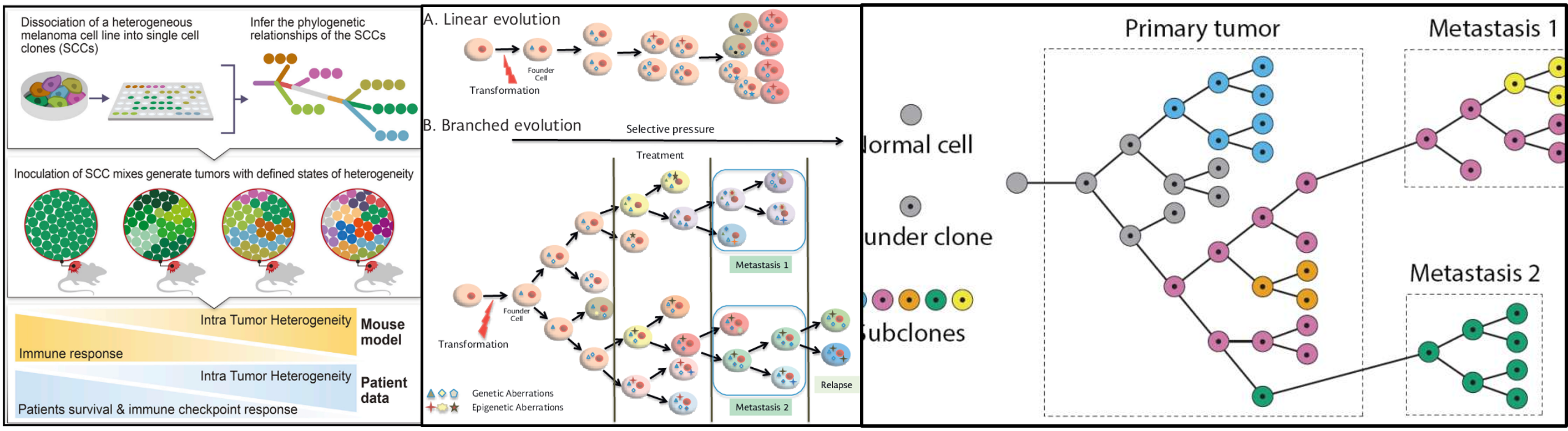
Recently, I need to give a talk about genetic and epigenetic based Intra-Tumor Heterogeneity (ITH)
Continue reading...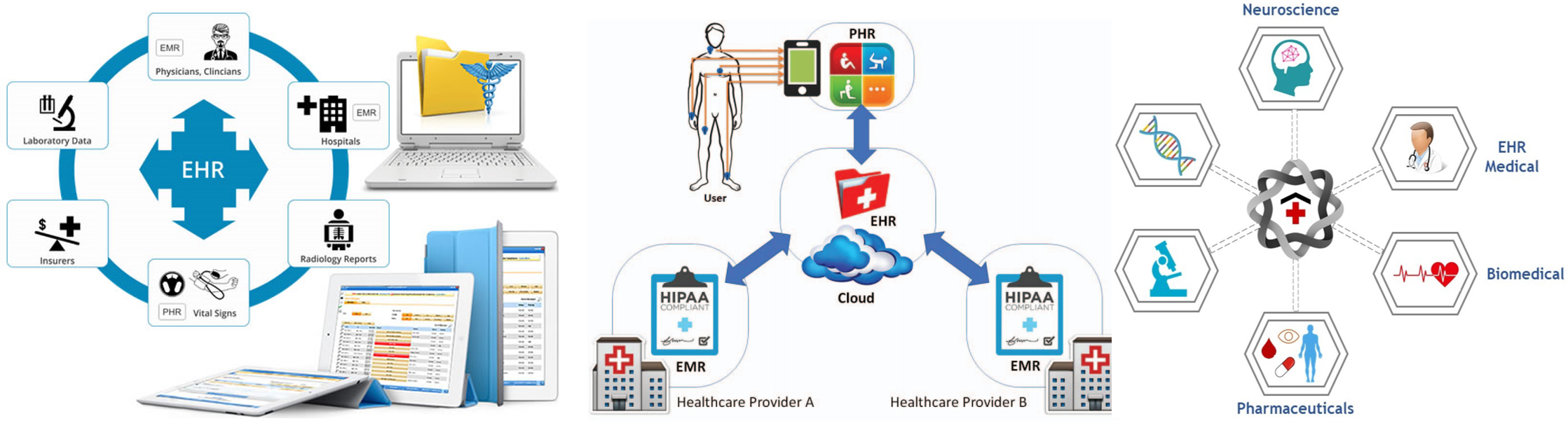
Recently, I need to give a talk about electronic health/medical records (EHR/EMR) data analysis. Why we need analyze EHR and EMR data? What’s the benefit and what’s the challenge? How to apply text-mining and data-mining in non-structural or semi-structual data analysis? How to deal with compliance and HIPPA? How to deal with multiple EMR systems and diffential format? the importance of genetic/genomic information in health care managment? the importance of the heatlh history of the patients? the mature pipeline to extract important clinical information? cancer real-world data (RWD) analysis? NCBI pubmed data re-leanring? How to connect bioinformatics team and data science as well as medical informatic scientist?
Continue reading...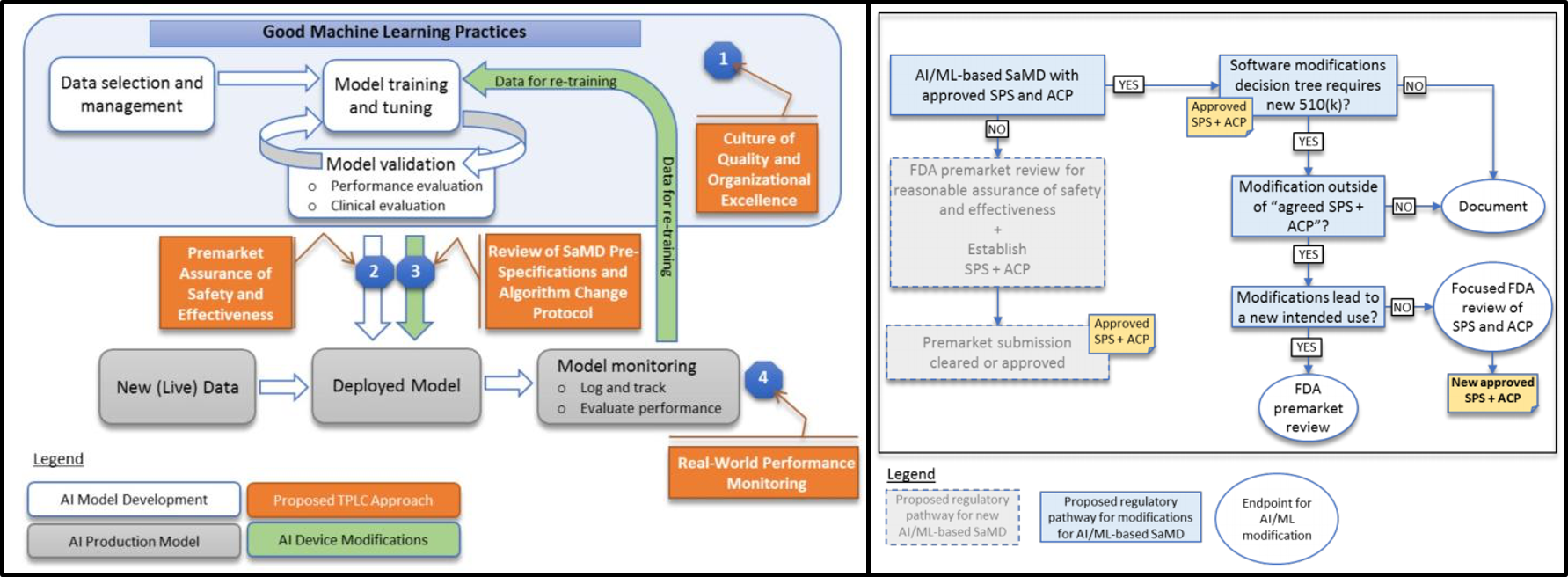
How to apply Artificial Intelligence in medical diagnosis and medical device from US-FDA policy? More and more patents based on artifical intelligence occured in medical diagnosis and medical service, however, FDA have its own policy in the usage of AI in medical production. Here, we discussed the best approach to design machine learning and AI in production strategy design. Usually, we will have training dataset and test dataset in the machine learning modeling. Here, I will use an example to introduce the best way to apply AI in your production design.
Continue reading...
“Artificial intelligence in risk prediction and mendelian diseases diagnosis”: Today, I want to give a talk about application of AI in risk prediction and disease diagnosis with deep sequencing and deep phenotyping. As the technique growth, we have already had the ability to generate deep sequencing data within short time, however, deep phenotyping is not easy work which limited the development of genetic association study. Actually, we can apply ** molecular phenotypes ** to replace “clinical or external phenotypes”.
Continue reading...
Today, I just submitted the letter of intent for the interal grant application in Marshfiled Clinic Research Institute as the PI. The proposal I submitted this year named: Deep Learning Prediction of Chemotherapy Response using Multi-Omics Features. Same with other grant, the first step of the application is submit letter of intent. You need prepare the title, PI/co-PIs, research abstract including background, hypothesis, aims, strategy and significance. You don’t need to get signature from all the co-PIs in this step. Usually the research abstract should be less than 500 words. You should submit 2 potential reviewer to review your abstract.
Continue reading...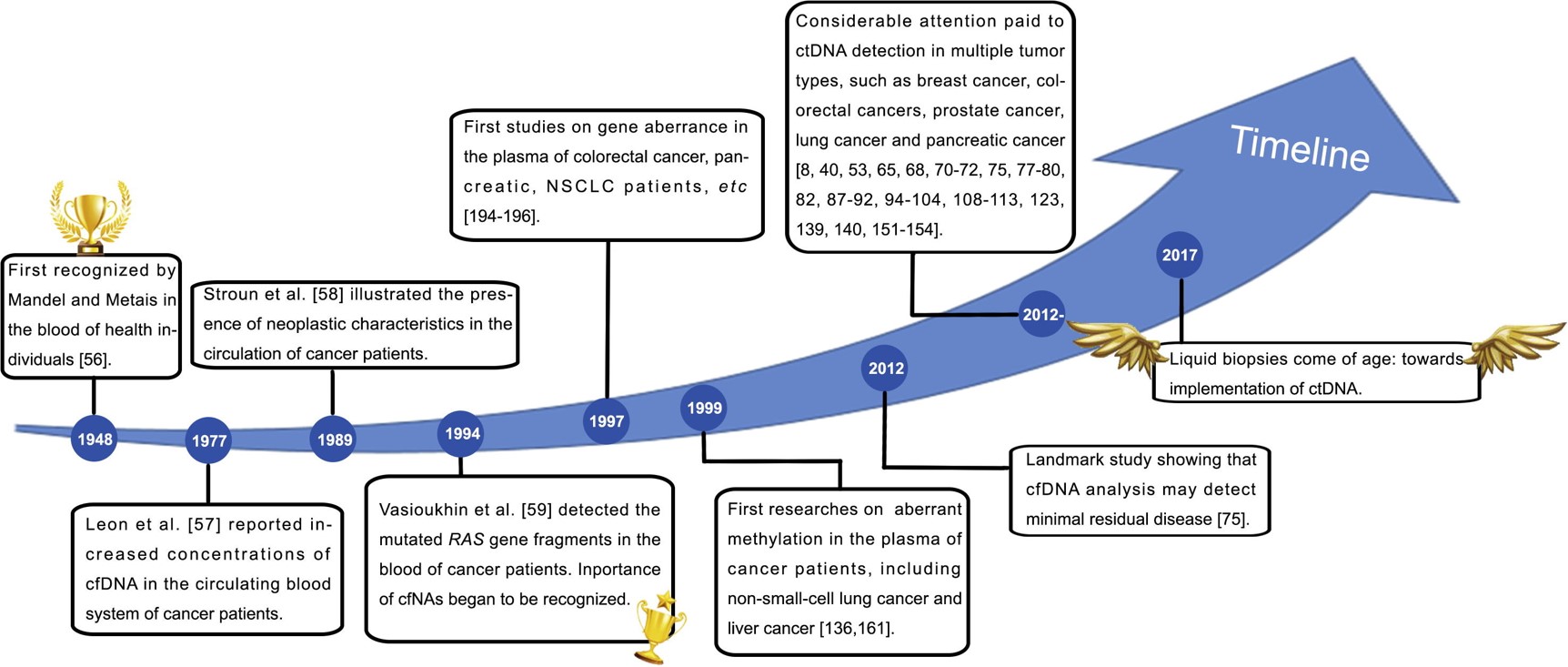
Today, I will give a talk about “Genome-wide cell-free DNA fragmentation in patients with cancer” in MCRI postdoc club meeting. The paper was published in Nature recently. See the slice here or download here for more details.
Continue reading...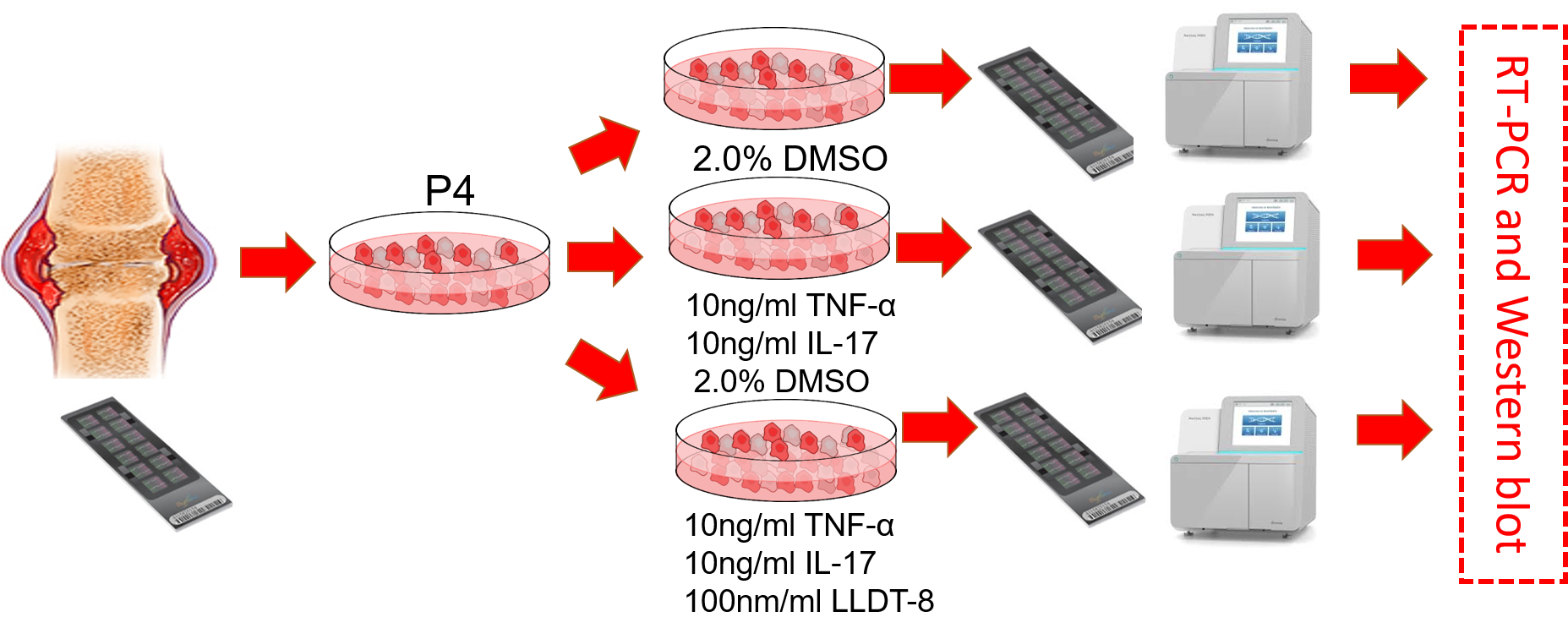
Recently, our project collaborated with Dr. Dongyi He in effects of (5R)-5-Hydroxytriptolide to epigenetic changes of LFS from RA was accepted by Scientfic Reports
Continue reading...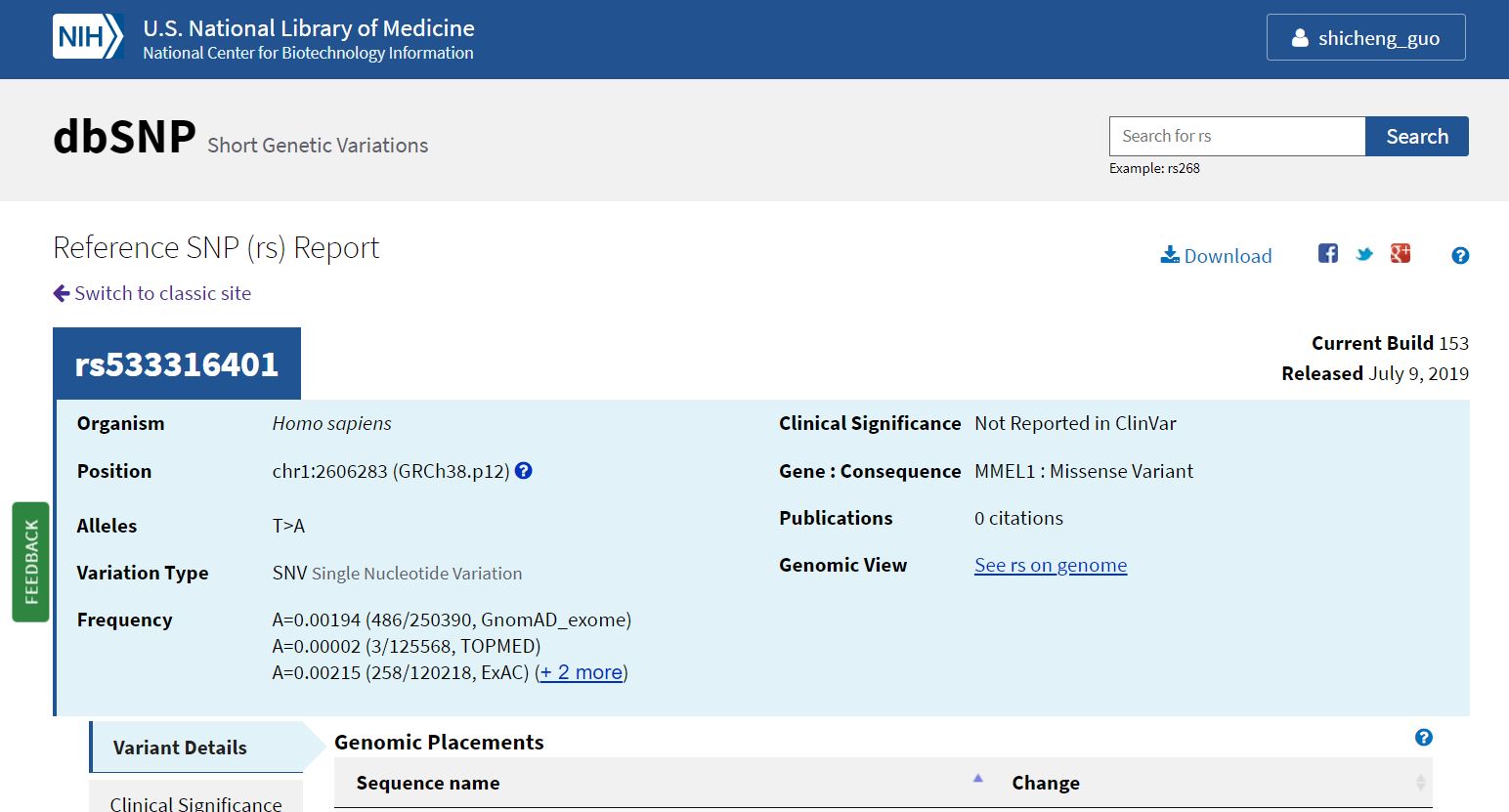
Recently (07/09/2019), dbSNP have been updated dbSNP153 from dbSNP152. However, NCBI only provided dbSNP153 in hg38 (GRCH38) version without any source for hg19 version. Here, I prepared a approach to generate dbSNP153 in hg19..
Continue reading...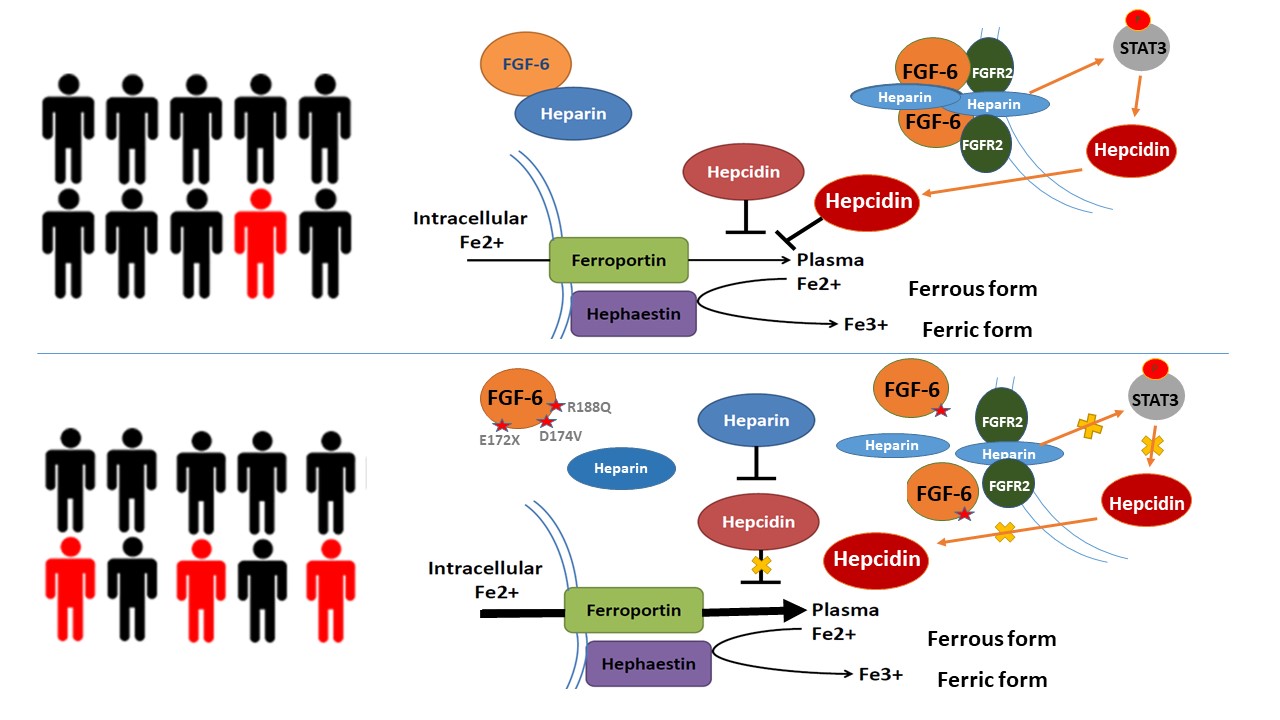
Recently, our project collaborated with Dr. Jiucun Wang named A gene-based recessive diplotype exome scan discovers FGF6, a novel hepcidin-regulating iron-metabolism gene was accepted by Blood
Continue reading...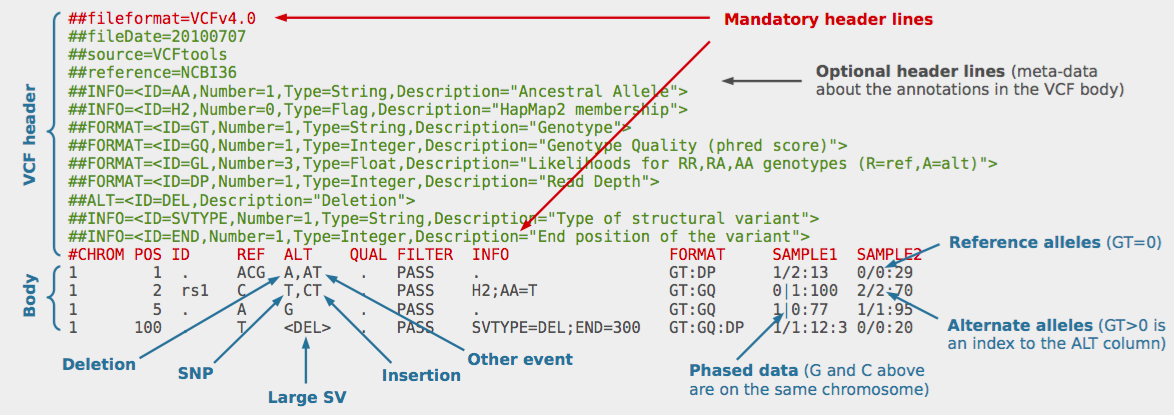
Today, I will give a talk about “Next generation protocol to bcftools in medical genetics research” in MCRI research hub meeting. As we know, bcftools, vcftools, plink2, GATK4 have been widely used in medical genetics and population genetics research. The usage of these tools require lots of experiences. However, the original protocols are quite limited espeically lacking of real-data example. Here, I will provide the real-data examples and solution to most frequently problem we meet in the usage of these tools.
Continue reading...
One of terrible things in marshfield clinic research institute (MCRI) is that software/package install request. Any software even Rstudio, R, Python will be reviewed by ITS department for security issues. Furthermore, the review process usually takes 2-3 months which make the research become quite difficult. Lucky thing is we can submit request as soon as possible supposing you think the software will be potentially used in the coming month. Finally, ITS don’t provide the software list which have been approved. therefore, it is quite necessary to list them and I think it will be helpful for further research fellows in MCRI.
Continue reading...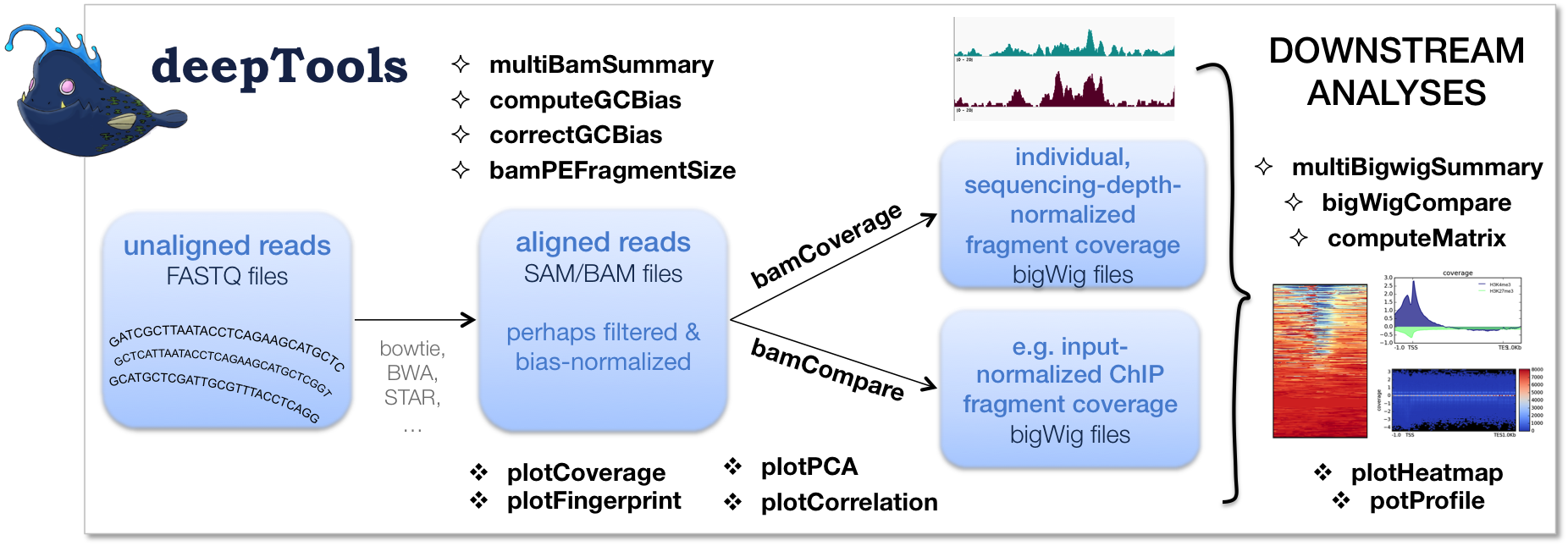
deepTools is a suite of python tools particularly developed for the efficient analysis of high-throughput sequencing data, such as ChIP-seq, RNA-seq or MNase-seq. deeptools has been widely applied in bam/bigwig data analysis. here, I show some example how to use deeptools in mbd-seq and medip-seq methylation data analysis. Meanwhile, MACS usage will also be shown in this poster.
Continue reading...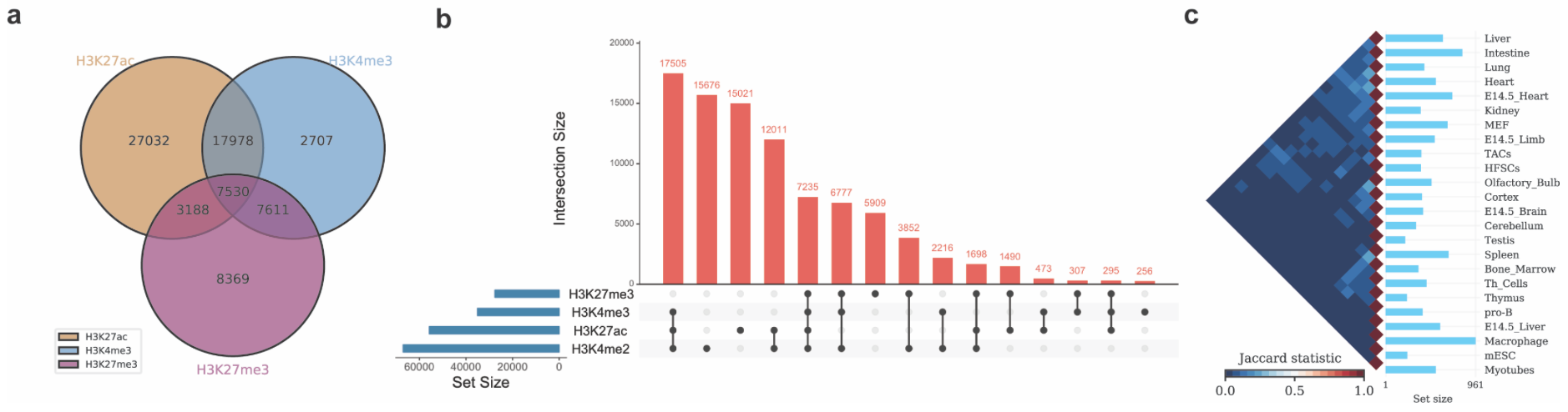
Today, I will give a talk about how to do multiple bedgraph data analysis with Intervene for ChIP-seq or MBD-seq data with Intervene. Intervene is a tool for intersection and visualization of multiple genomic region and gene sets (or lists of items). Intervene provides an easy and automated interface for effective intersection and visualization of genomic region sets or lists of items, thus facilitating their analysis and interpretations.
Continue reading...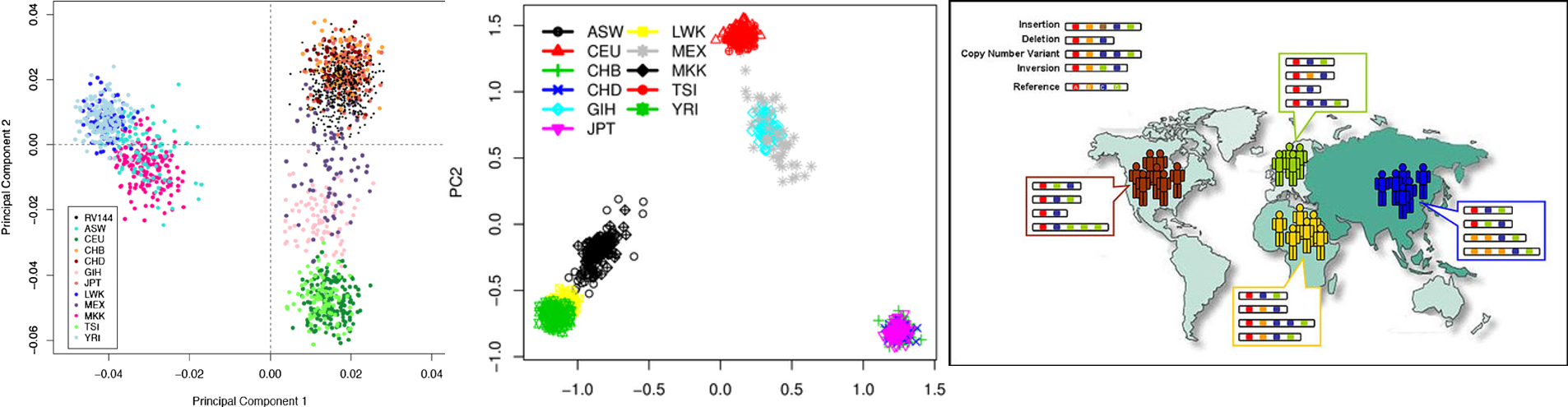
Recently, colleagues always ask me what’s the best solution to update hapmap3 from hg18 to hg19 and hg38. Here, I try to give certain solutions.
Continue reading...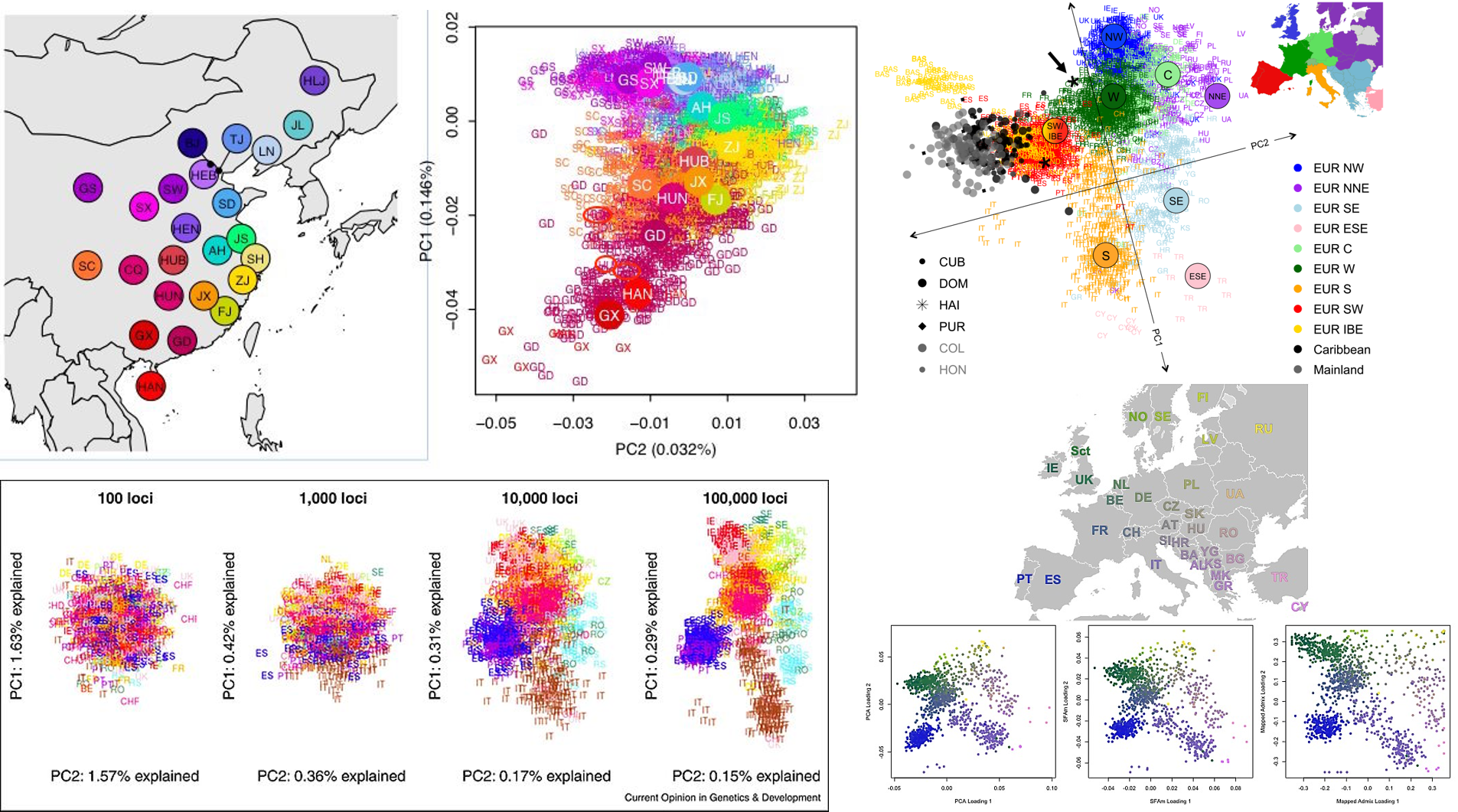
Here, Characterizing population with principle componment analysis
Continue reading...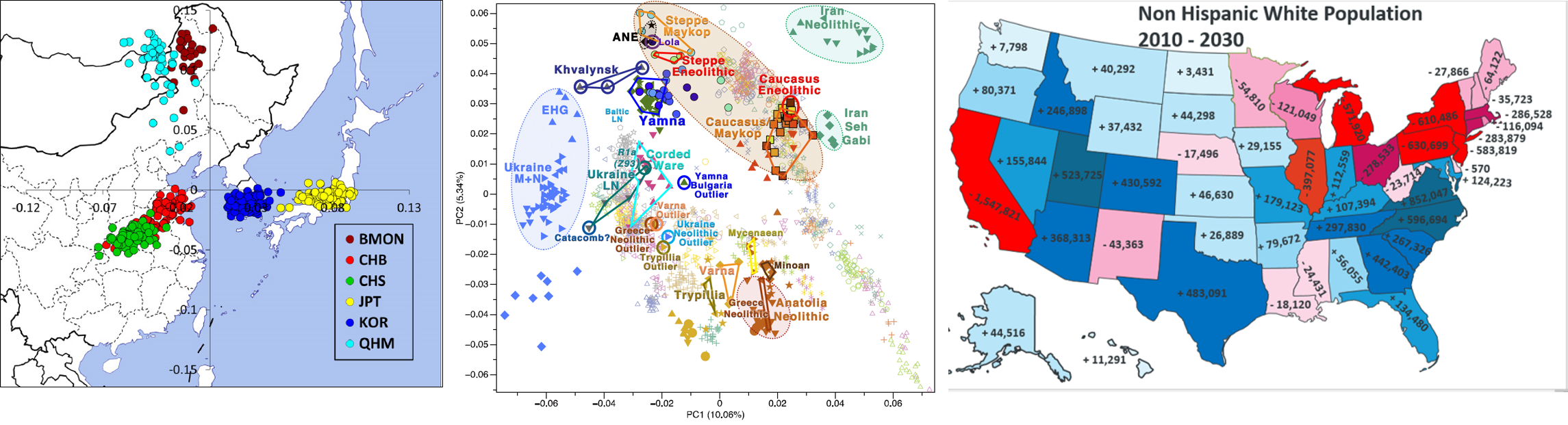
Here, Data Science in Population Genetics and Medical Genetics
Continue reading...
Here, I want to summarized the genetic and epigeneitc difference between rheumatoid arthritis(RA) and osteoarthritis(OA). OA and RA have certain similarity such as joint demage..Genetics/Epigenetics
Continue reading...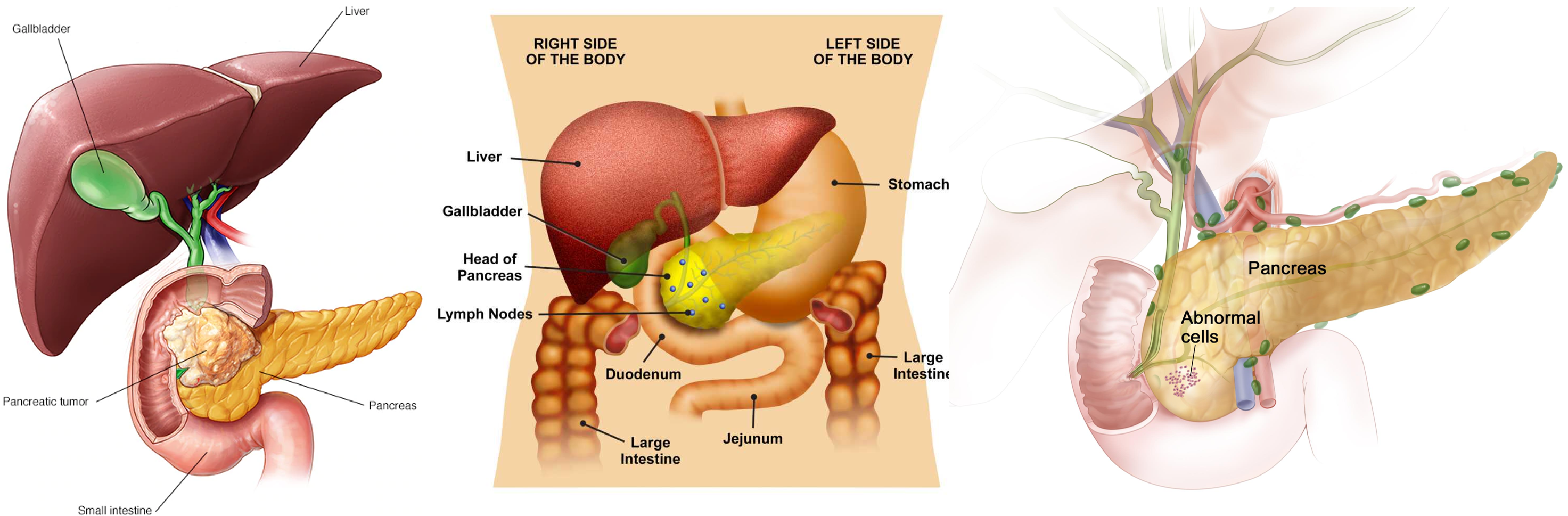
{kind=link}
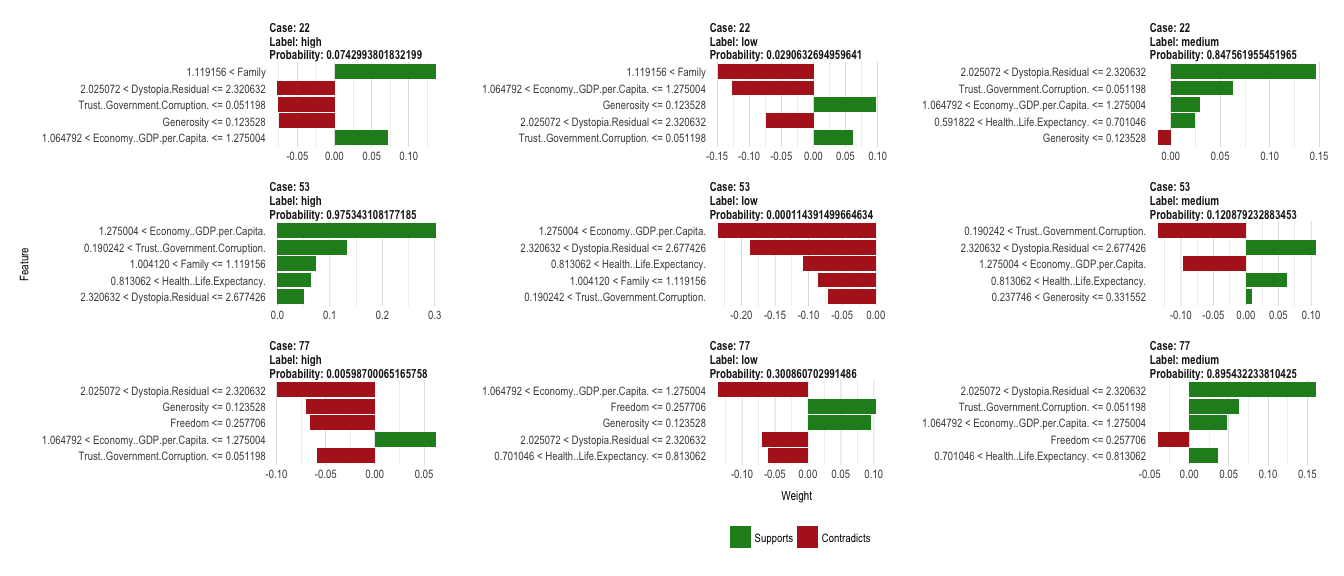
The classification decisions made by machine learning models are usually difficult - if not impossible - to understand by our human brains. The complexity of some of the most accurate classifiers, like neural networks, is what makes them perform so well - often with better results than achieved by humans. But it also makes them inherently hard to explain, especially to non-data scientists.
Continue reading...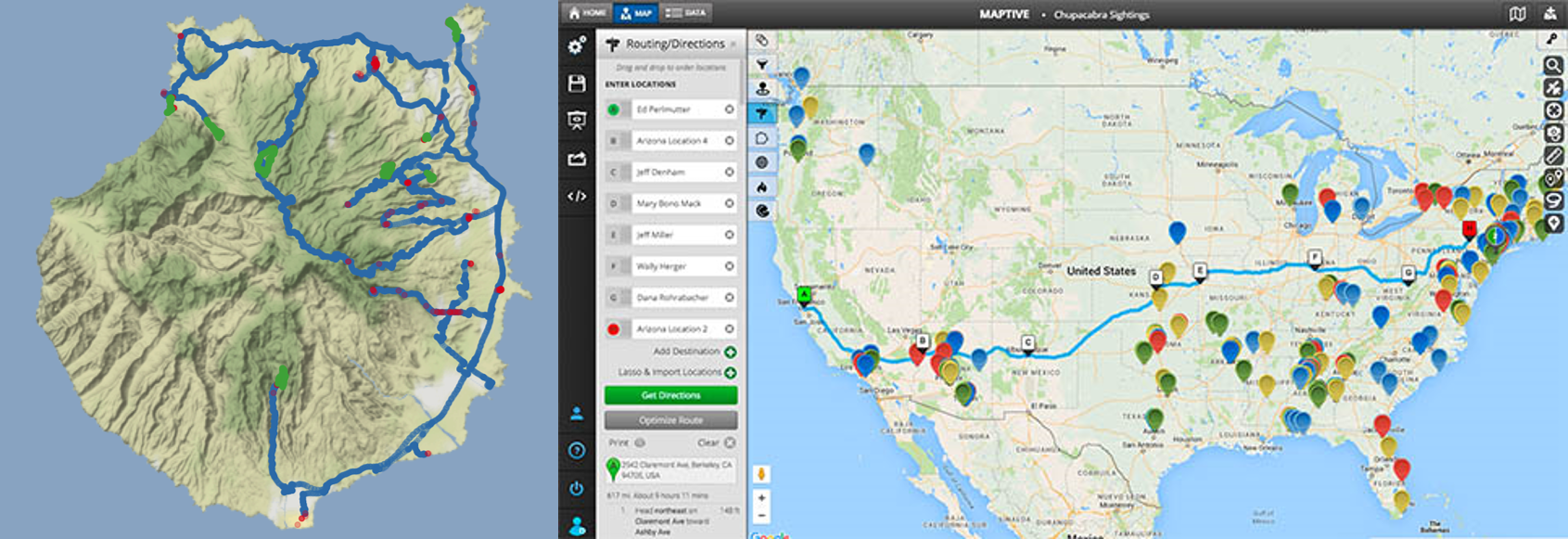
Recently, I was on Gran Canaria for a vacation. So, what better way to keep up the holiday spirit a while longer than to visualize all the places we went in R!?
Continue reading...
Science Writing: Guidelines And Guidance
Continue reading...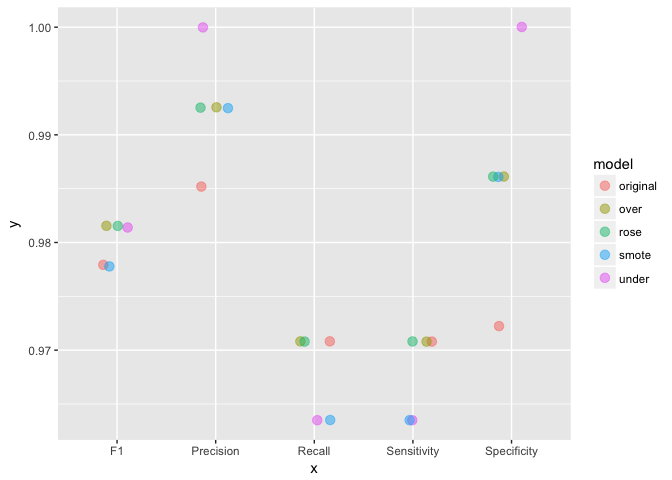
In my last post, where I shared the code that I used to produce an example analysis to go along with my webinar on building meaningful models for disease prediction, I mentioned that it is advised to consider over- or under-sampling when you have unbalanced data sets. Because my focus in this webinar was on evaluating model performance, I did not want to add an additional layer of complexity and therefore did not further discuss how to specifically deal with unbalanced data.
Continue reading...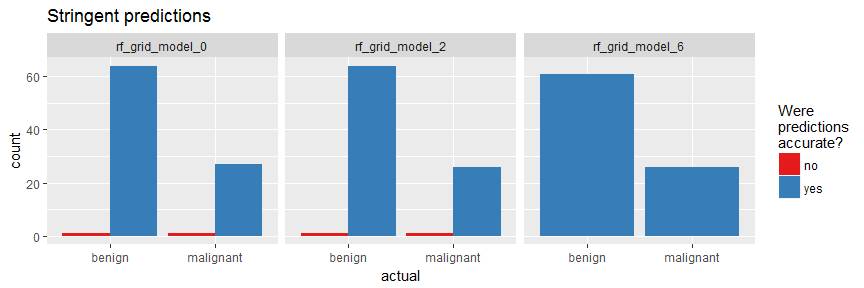
Webinar for the ISDS R Group
Continue reading...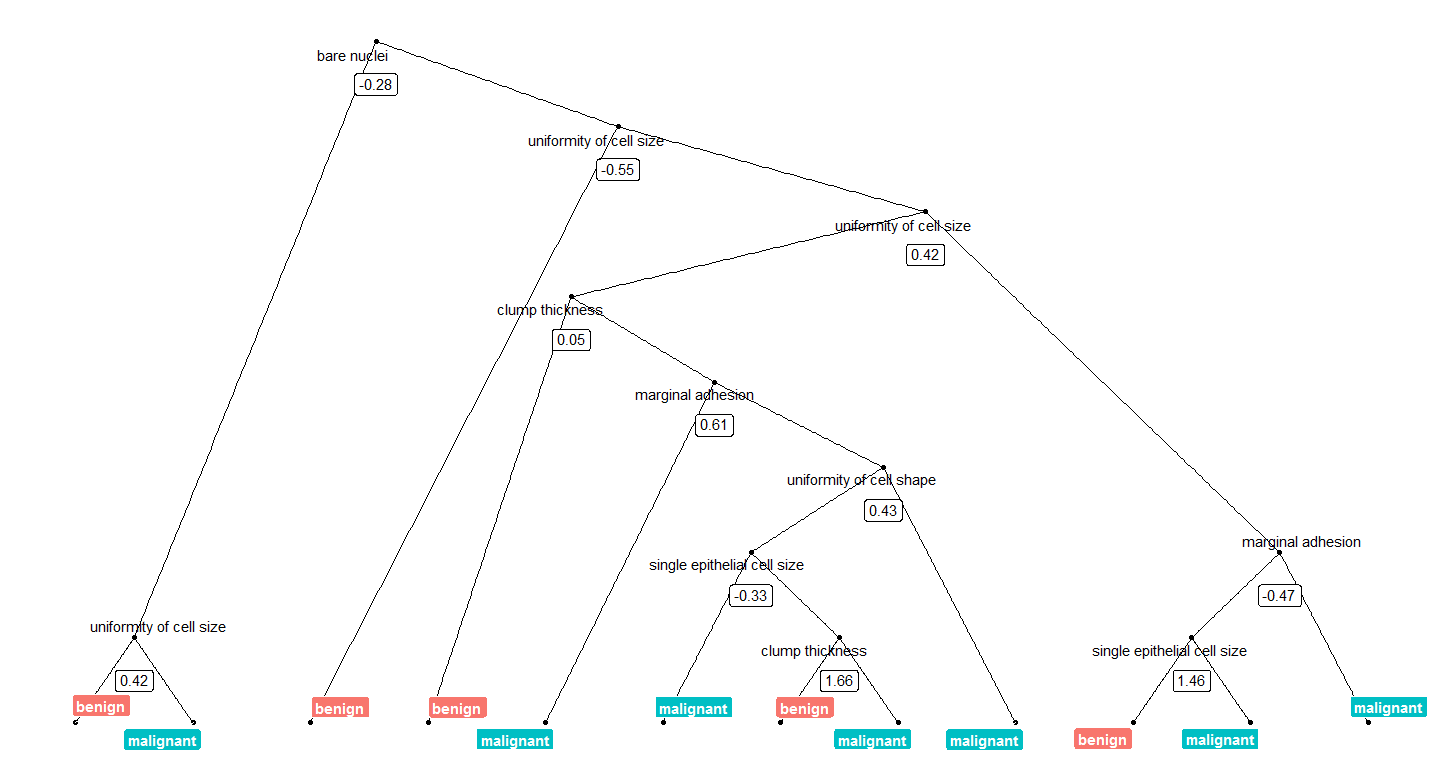
Today, I want to show how I use Thomas Lin Pedersen’s awesome ggraph package to plot decision trees from Random Forest models.
Continue reading...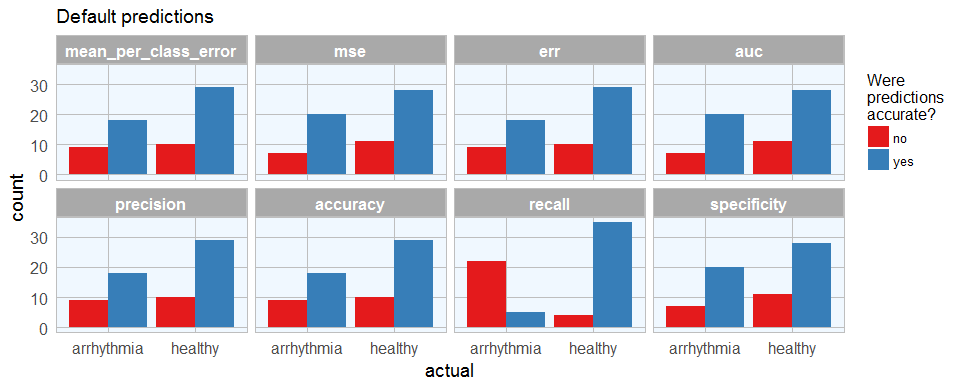
Last week I showed how to build a deep neural network with h2o and rsparkling. As we could see there, it is not trivial to optimize the hyper-parameters for modeling. Hyper-parameter tuning with grid search allows us to test different combinations of hyper-parameters and find one with improved accuracy.
Continue reading...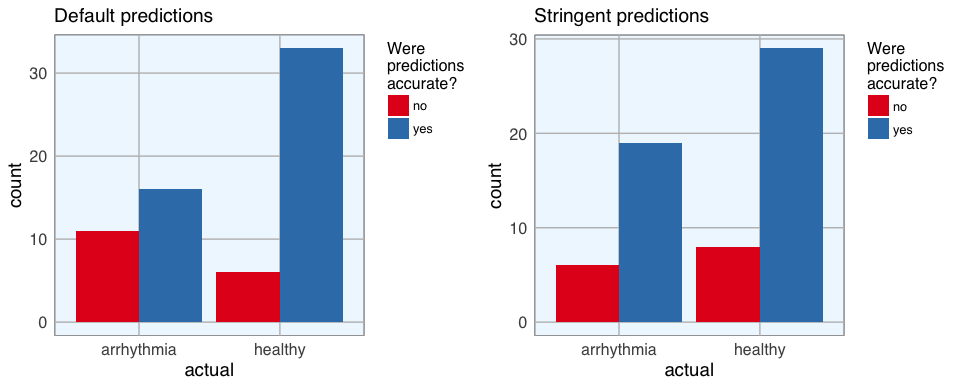
Last week, I introduced how to run machine learning applications on Spark from within R, using the sparklyr package. This week, I am showing how to build feed-forward deep neural networks or multilayer perceptrons. The models in this example are built to classify ECG data into being either from healthy hearts or from someone suffering from arrhythmia. I will show how to prepare a dataset for modeling, setting weights and other modeling parameters and finally, how to evaluate model performance with the h2o package via rsparkling.
Continue reading...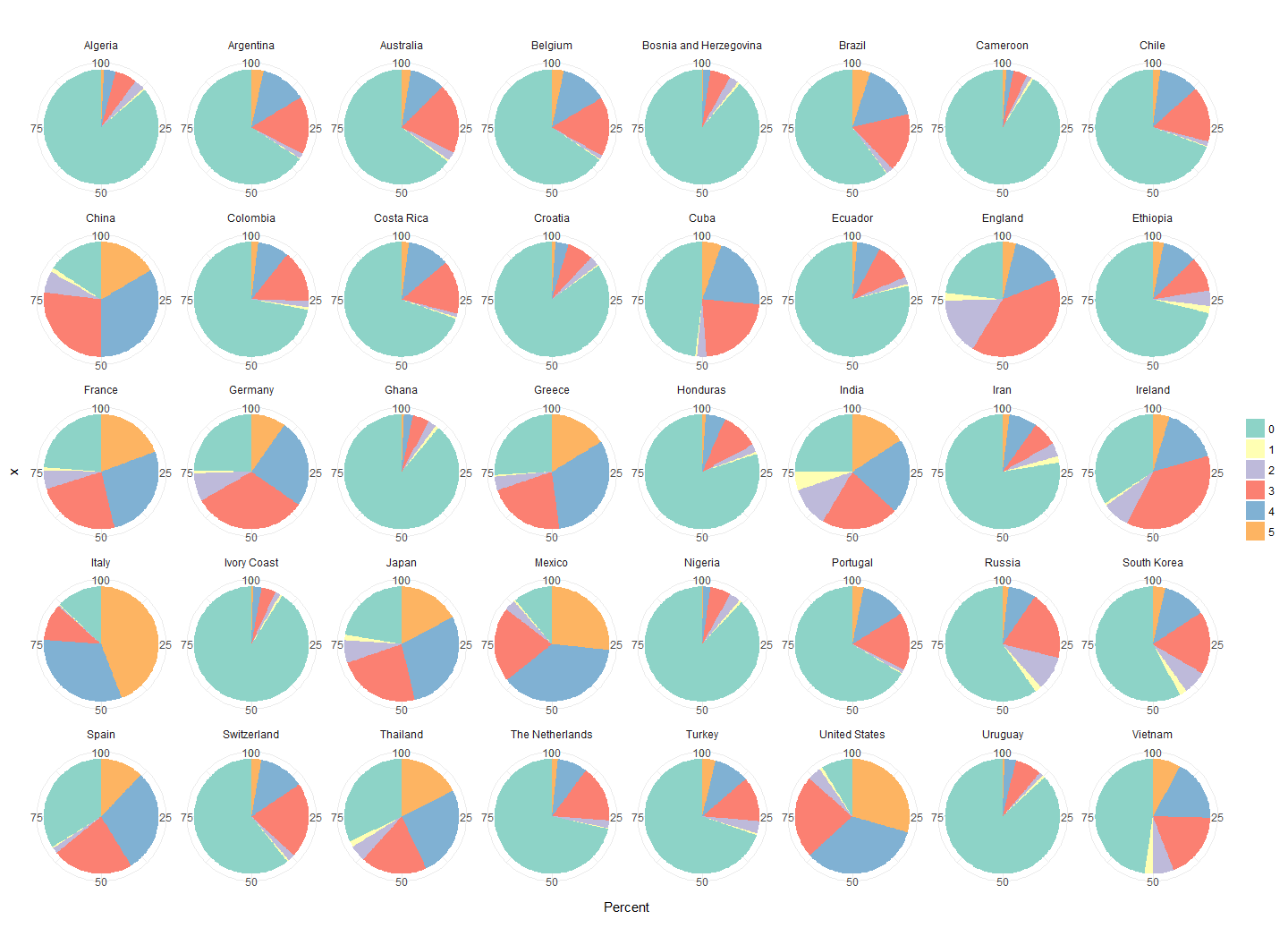
This week I want to show how to run machine learning applications on a Spark cluster. I am using the sparklyr package, which provides a handy interface to access Apache Spark functionalities via R.
Continue reading...
When running an analysis, I am usually combining functions from multiple packages. Most of these packages come with their own plotting functions. And while they are certainly convenient in that they allow me to get a quick glance at the data or the output, they all have their own style. If I want to prepare a report, proposal or a paper though, I want all my plots to come from a single cast so that they give a consistent feel to the story I want to tell with my data.
Continue reading...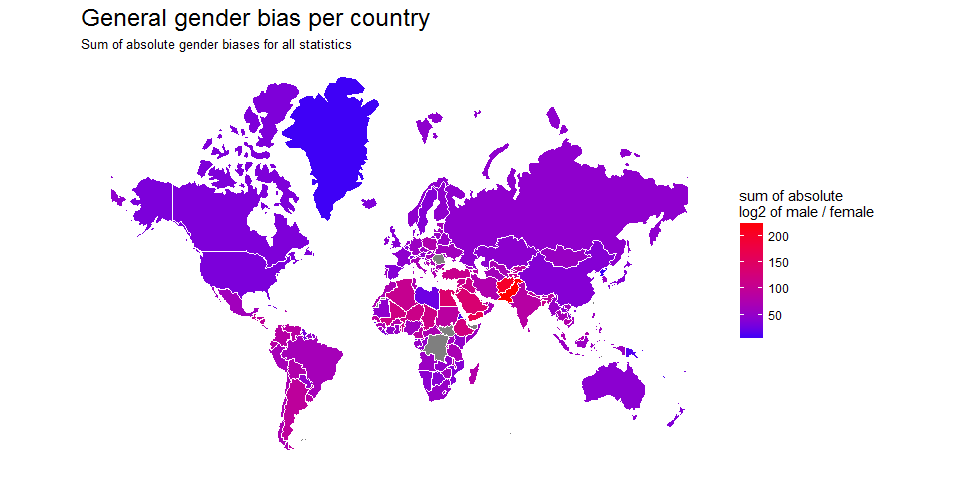
Today, I want to share my analysis of the World Gender Statistics dataset.
Continue reading...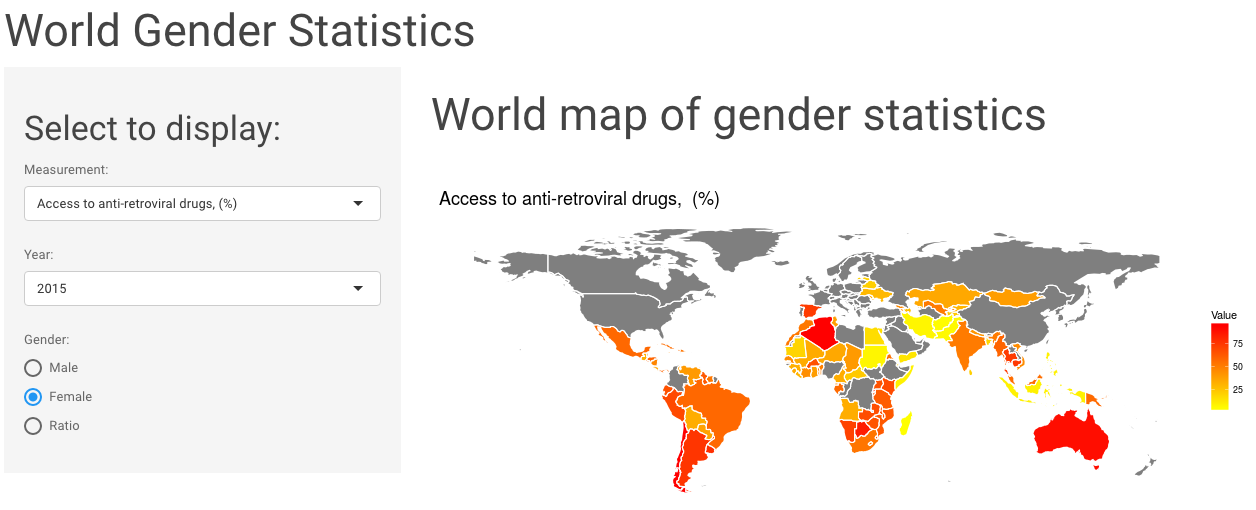
In my last post, I built a shiny app to explore World Gender Statistics.
Continue reading...
This week I explored the World Gender Statistics dataset. You can look at 160 measurements over 56 years with my Shiny app here.
Continue reading...
I’m an avid R user and rarely use anything else for data analysis and visualisations. But while R is my go-to, in some cases, Python might actually be a better alternative.
Continue reading...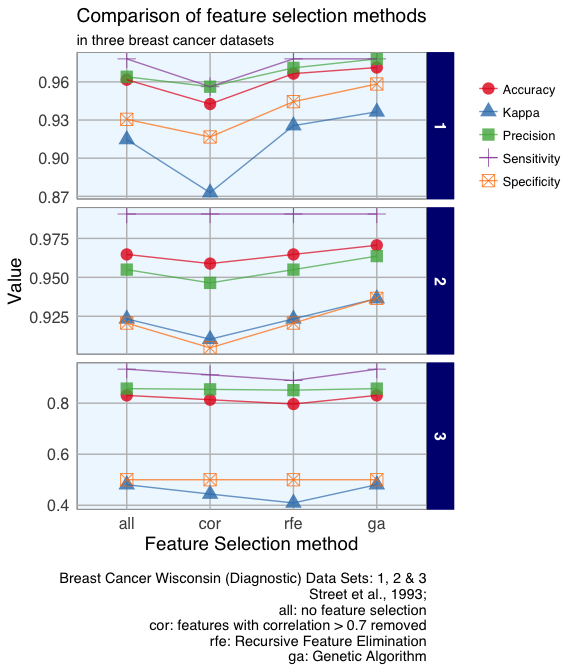
Machine learning uses so called features (i.e. variables or attributes) to generate predictive models. Using a suitable combination of features is essential for obtaining high precision and accuracy. Because too many (unspecific) features pose the problem of overfitting the model, we generally want to restrict the features in our models to those, that are most relevant for the response variable we want to predict. Using as few features as possible will also reduce the complexity of our models, which means it needs less time and computer power to run and is easier to understand.
Continue reading...
Which genes have homologs in many species?
Continue reading...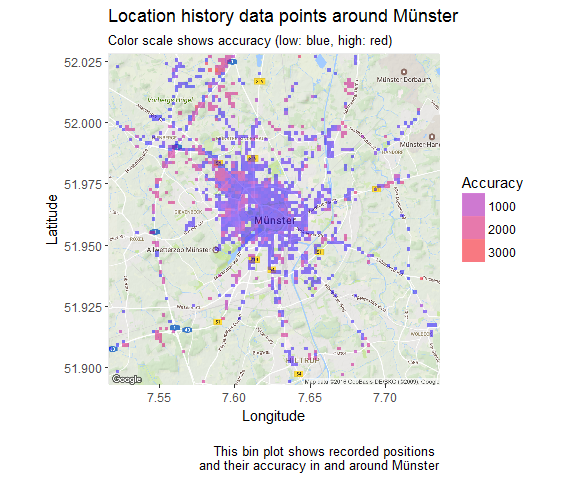
It’s no secret that Google Big Brothers most of us. But at least they allow us to access quite a lot of the data they have collected on us. Among this is the Google location history.
Continue reading...
With the upcoming holidays, I thought it fitting to finally explore the ttbbeer package. It contains data on beer ingredients used in US breweries from 2006 to 2015 and on the (sin) tax rates for beer, champagne, distilled spirits, wine and various tobacco items since 1862.
Continue reading...
This app is based on the gwascat R package and its ebicat38 database and shows trait-associated SNP locations of the human genome. You can visualize and compare the genomic locations of up to 8 traits simultaneously.
Continue reading...
In my last post I created a gene homology network for human genes. In this post I want to extend the network to include edges for other species.
Continue reading...
Edited on 20 December 2016
Continue reading...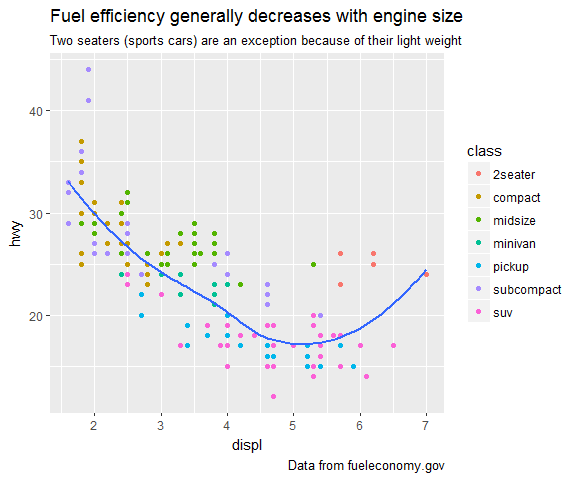
This post is in reply to a request: How did I set up this R blog?
Continue reading...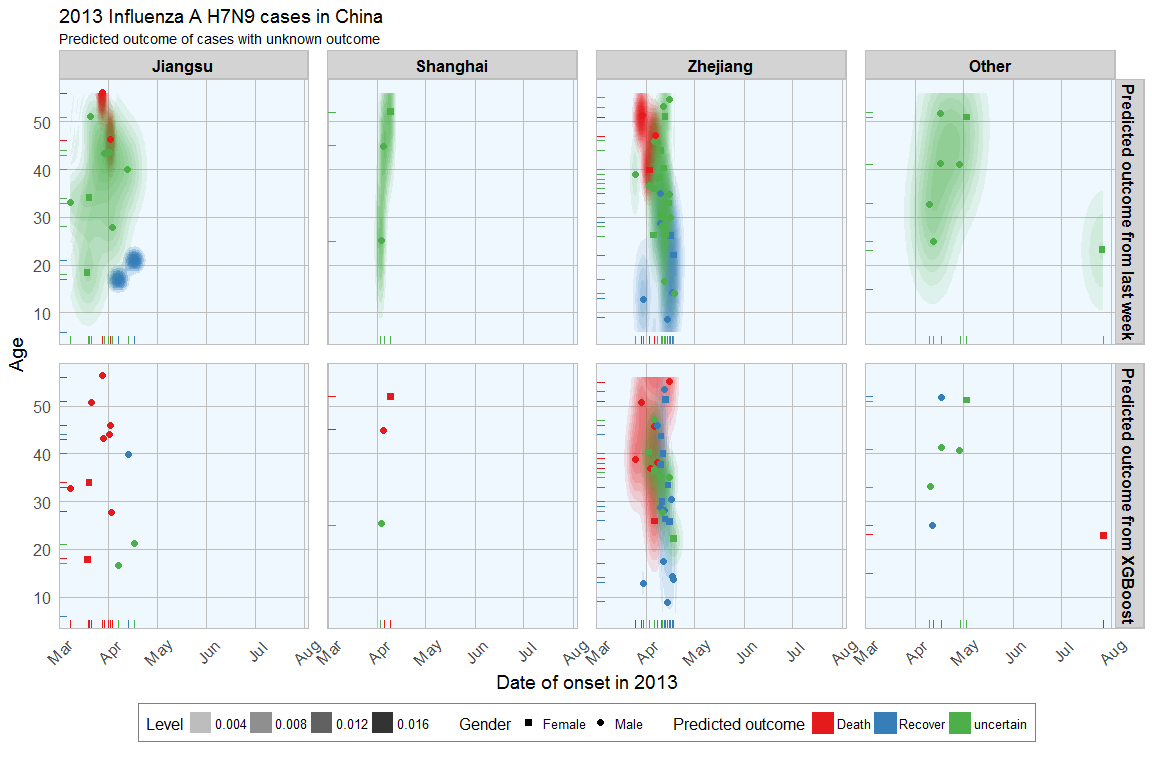
In last week’s post I explored whether machine learning models can be applied to predict flu deaths from the 2013 outbreak of influenza A H7N9 in China. There, I compared random forests, elastic-net regularized generalized linear models, k-nearest neighbors, penalized discriminant analysis, stabilized linear discriminant analysis, nearest shrunken centroids, single C5.0 tree and partial least squares.
Continue reading...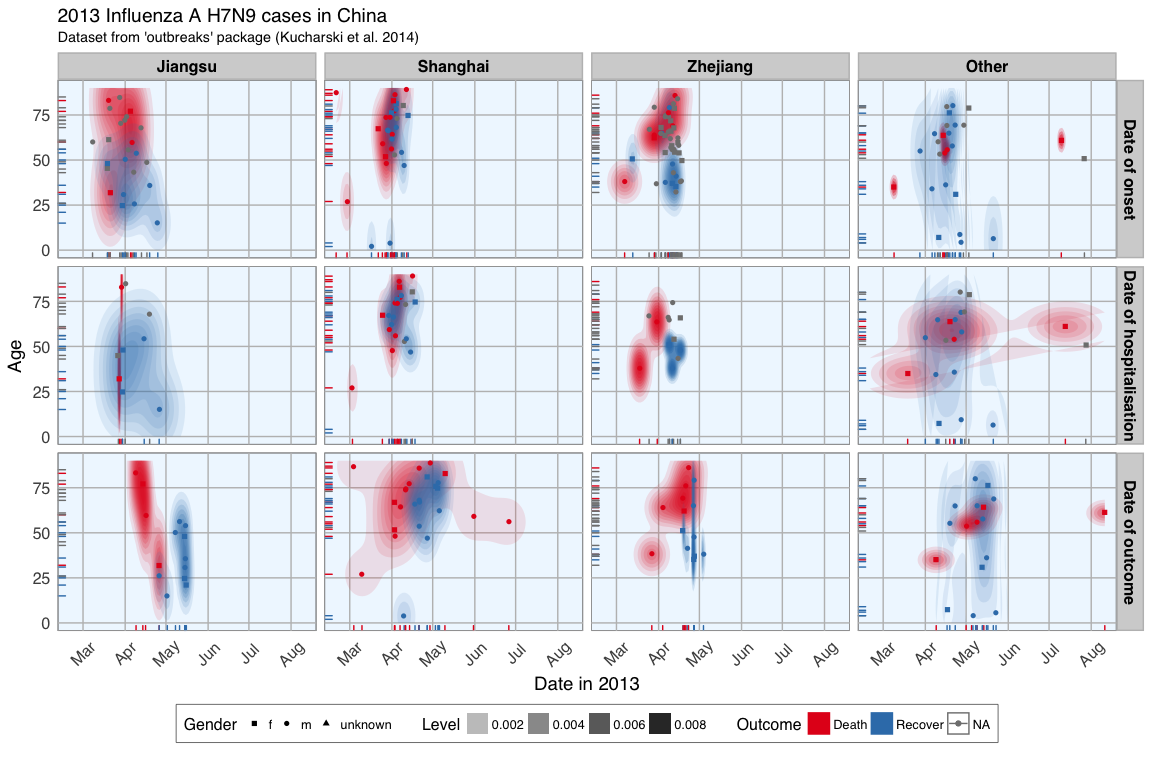
Edited on 26 December 2016
Continue reading...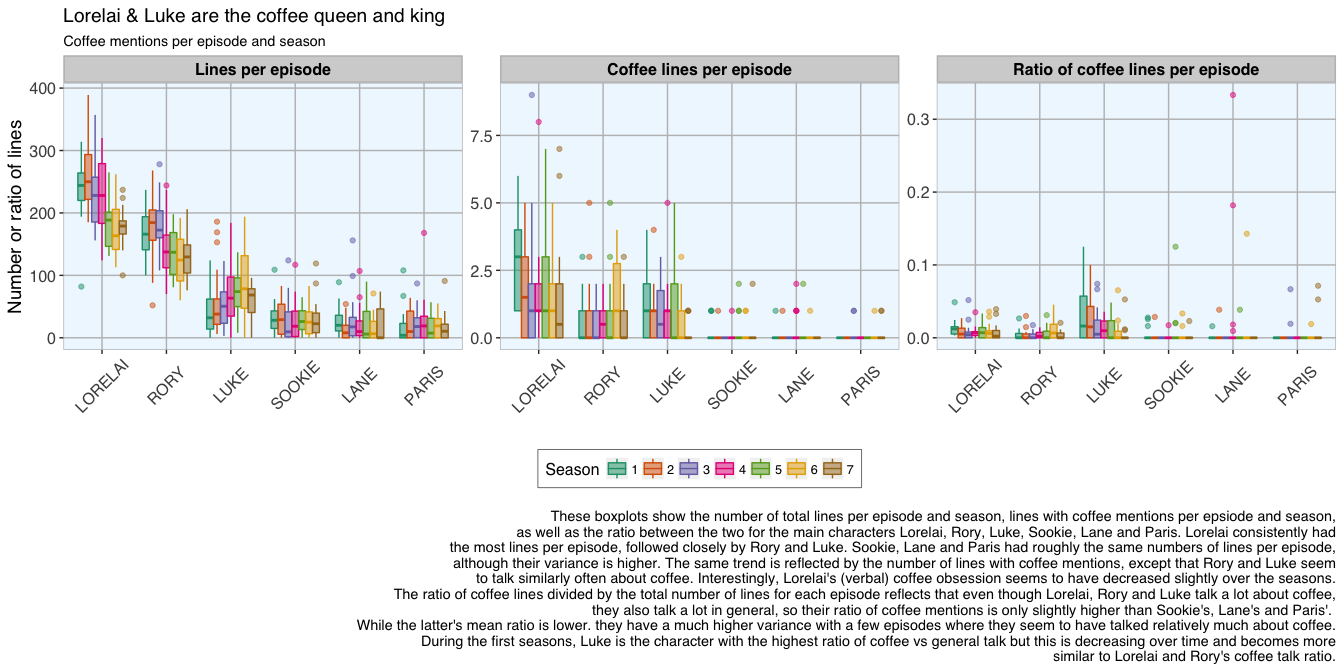
Last week’s post showed how to create a Gilmore Girls character network.
Continue reading...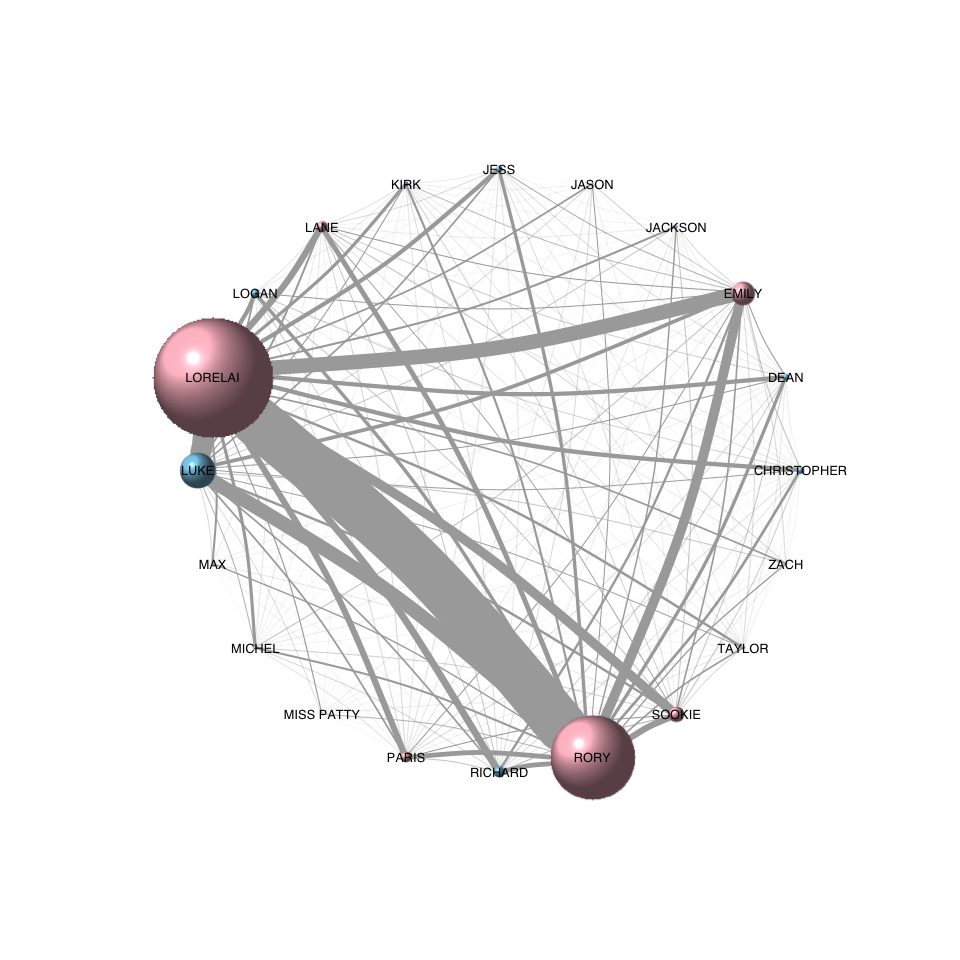
With the impending (and by many - including me - much awaited) Gilmore Girls Revival, I wanted to take a somewhat different look at our beloved characters from Stars Hollow.
Continue reading...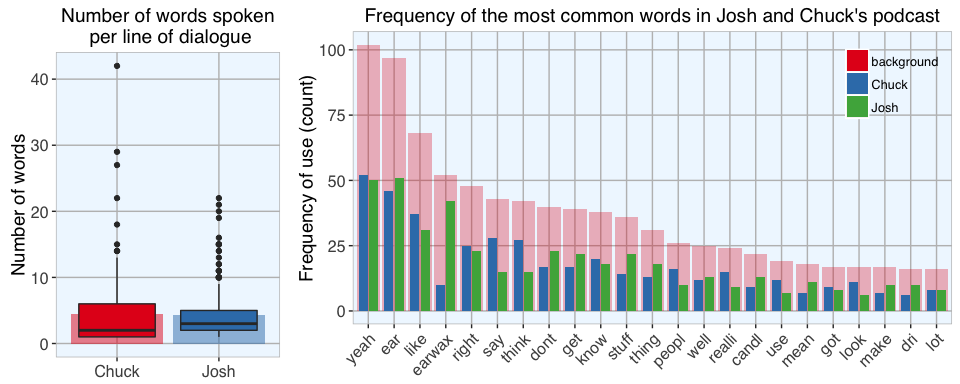
Text mining and sentiment analysis of a Stuff You Should Know Podcast
Continue reading...
How many transcripts and proteins do genes have?
Continue reading...
When working with any type of genome data, we often look for annotation information about genes, e.g. what’s the gene’s full name, what’s its abbreviated symbol, what ID it has in other databases, what functions have been described, how many and which transcripts exist, etc.
Continue reading...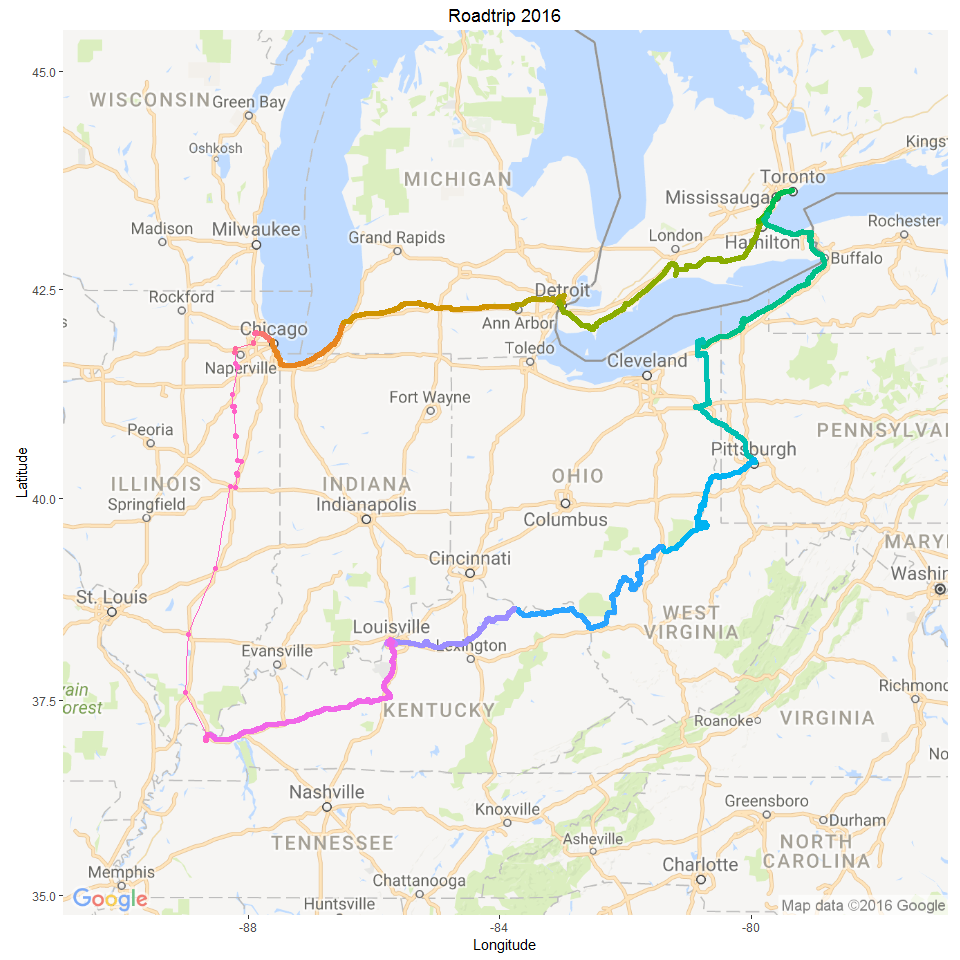
Mapping GPS data from our USA/ Canada Roadtrip
Continue reading...
Continue reading...
I created the R package exprAnalysis designed to streamline my RNA-seq data analysis pipeline. Below you find the vignette for installation and usage of the package.
Continue reading...
This post is to record all the environment setting for my previous work stations. the best way should be record the installation for all these tools, however, they are always being updated so here I only record them.
Continue reading...
The current post want to use STAR and HTseq together to estimation gene expression for RNA-seq:
Continue reading...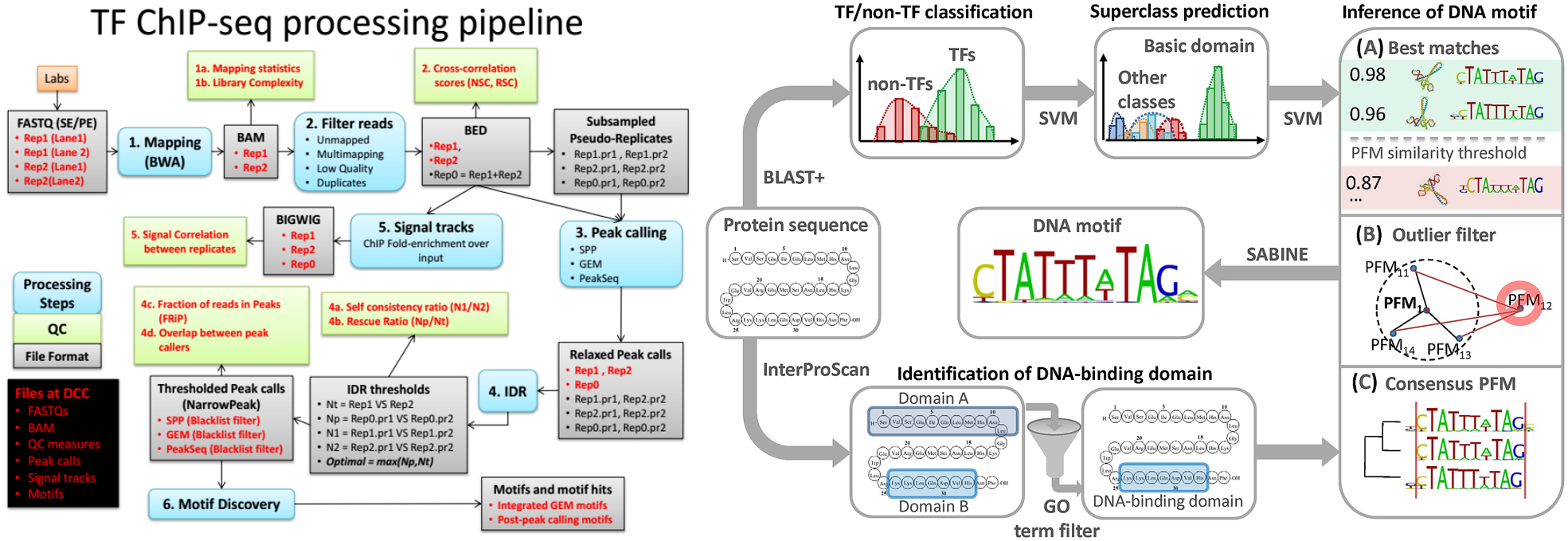
BooK
Continue reading...
DNA methylation
Continue reading...
Protocol for DNA demethylation with 5-aza-2’-deoxycytidine (DAC) treatment
Continue reading...
How to apply RIblast to predict ncRNA interaction target ``` cd ~/hpc/tools/RIblast/extdata wget http://ftp.ensembl.org/pub/release-97/fasta/homo_sapiens/ncrna/Homo_sapiens.GRCh38.ncrna.fa.gz wget http://ftp.ensembl.org/pub/release-97/fasta/homo_sapiens/cdna/Homo_sapiens.GRCh38.cdna.all.fa.gz wget http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/mrna.fa.gz -O Homo_sapiens.GRCh38.mrna.fa.gz wget ftp://ftp.ebi.ac.uk/pub/databases/gencode/Gencode_human/release_31/gencode.v31.transcripts.fa.gz
Continue reading...
Here is the best solution:
Continue reading...
Immune escape mechanisms
Continue reading...
Step 1: creat Bioproject (receive PRJNA ID to be used in step 2: PRJNA605250)
Continue reading...
Resources for flow cytometry bioinformatics analysis
Continue reading...
how do I prune second-degree-related samples? —rel-cutoff is obsolete and see —king-cutoff
plink --bfile RA1000 --bmerge RA500 --make-bed --out RA3000
plink --bfile RA3000 --impute-sex --make-bed --out RA3000.R1
grep PROBLEM RA3000.R1.sexcheck | awk '{print $2}' > sexcheck.exclude.txt
plink --bfile RA3000 --impute-sex --exclude --make-bed --out RA3000.R1
plink2 —bfile ... —king-cutoff 0.088 --maf 0.01 —make-bed --out myplink
Compare plink bim to 1000G/HRC dataset
```
Script to check plink .bim files against HRC/1000G for strand, id names, positions, alleles,
ref/alt assignment, William Rayner 2015, wrayner@well.ox.ac.uk, Version 4.2.7
cd /mnt/sas0/AD/sguo234/asa wget http://www.well.ox.ac.uk/~wrayner/tools/HRC-1000G-check-bim-v4.2.7.zip wget http://ngs.sanger.ac.uk/production/hrc/HRC.r1-1/HRC.r1-1.GRCh37.wgs.mac5.sites.tab.gz wget https://www.well.ox.ac.uk/~wrayner/tools/1000GP_Phase3_combined.legend.gz gunzip 1000GP_Phase3_combined.legend.gz unzip HRC-1000G-check-bim-v4.2.7.zip gunzip HRC.r1-1.GRCh37.wgs.mac5.sites.tab.gz wget http://qbrc.swmed.edu/zhanxw/software/checkVCF/checkVCF-20140116.tar.gz tar xzvf checkVCF-20140116.tar.gz
compare bim to HRC European Population(HEP)
perl HRC-1000G-check-bim.pl -b RA3000.R3.bim -f RA3000.R3.frq -r HRC.r1-1.GRCh37.wgs.mac5.sites.tab -h sh Run-plink.sh
compare bim to 1000G East-Asian Population(EAP)
perl HRC-1000G-check-bim.pl -b RA3000.R3.bim -f RA3000.R3.frq -r 1000GP_Phase3_combined.legend -g -p EAS sh Run-plink.sh
How to add rs ID to replace illumina array probes (exm) with bcf annotate (use -m-both to extent multi-allic SNPs to multiple row)
Continue reading...
bacterial RNA-seq analysis with Rockhopper: RNA-seq data to reveal novel response mechanism to bacterial within host wound tissues
Continue reading...
Script A: transfer final report to ped (fr2ped.pl)
use strict;
use Cwd;
chdir getcwd;
open F,shift @ARGV;
my $i=1;
while(<F>){
next if !/ZS/;
my($snp,$sam,$rs,$gc,$chr,$pos,$a1,$a2,undef)=split/\s+/;
if($i eq 1){
print "$sam $sam 0 0 0 0 $a1 $a2";
}else{
print " $a1 $a2";
}
$i++;
}
print "\n";
Script B: transfer final report to ped(fr2map.pl)
use strict;
use Cwd;
chdir getcwd;
open F,shift @ARGV;
while(<F>){
next if !/ZS/;
my($snp,$sam,$rs,$gc,$chr,$pos,$a1,$a2,undef)=split/\s+/;
print "$chr $rs 0 $pos\n";
}
Step 3.0: run the script to do the job
```
for i in ls *.txt | rev | cut -c 17- | rev | uniq
do
echo $i
perl ./fr2ped.pl $i_FinalReport.txt > $i.ped
done
Sometimes, maybe you want to merge >7000 vcf files/samples into one big VCF file with bcftools merge, for example PMRP have 20,000 samples/vcf files:
bcftools merge -l merge.txt -Oz -o merge.vcf.gz
if the sample counts <1021, everything is okay. However, if it is >= 1021, bcftools merge will reports:
[E::hts_idx_load3] Could not load local index file '229209.fstl1.vcf.gz.tbi'
Failed to open 229209.fstl1.vcf.gz: could not load index
Okay. Here is my final solution developed based on WouterDeCoste’s post. I hope it is helpful. One of my friends told me his computer allowed merging 7000 VCF at one time. I am not sure whether it is caused by a specific file operating setting. ``` ls *.vcf.gz | split -l 500 - subset_vcfs
Continue reading...
Here, I summarized Automatic GWAS and Post-GWAS Analysis Pipeline Published Works:
Continue reading...
1, download ANNOVAR: http://annovar.openbioinformatics.org/en/latest/user-guide/download/
Continue reading...
Here, I list all the used annotation in my previous publication:
Continue reading...
Plan-A
Continue reading...
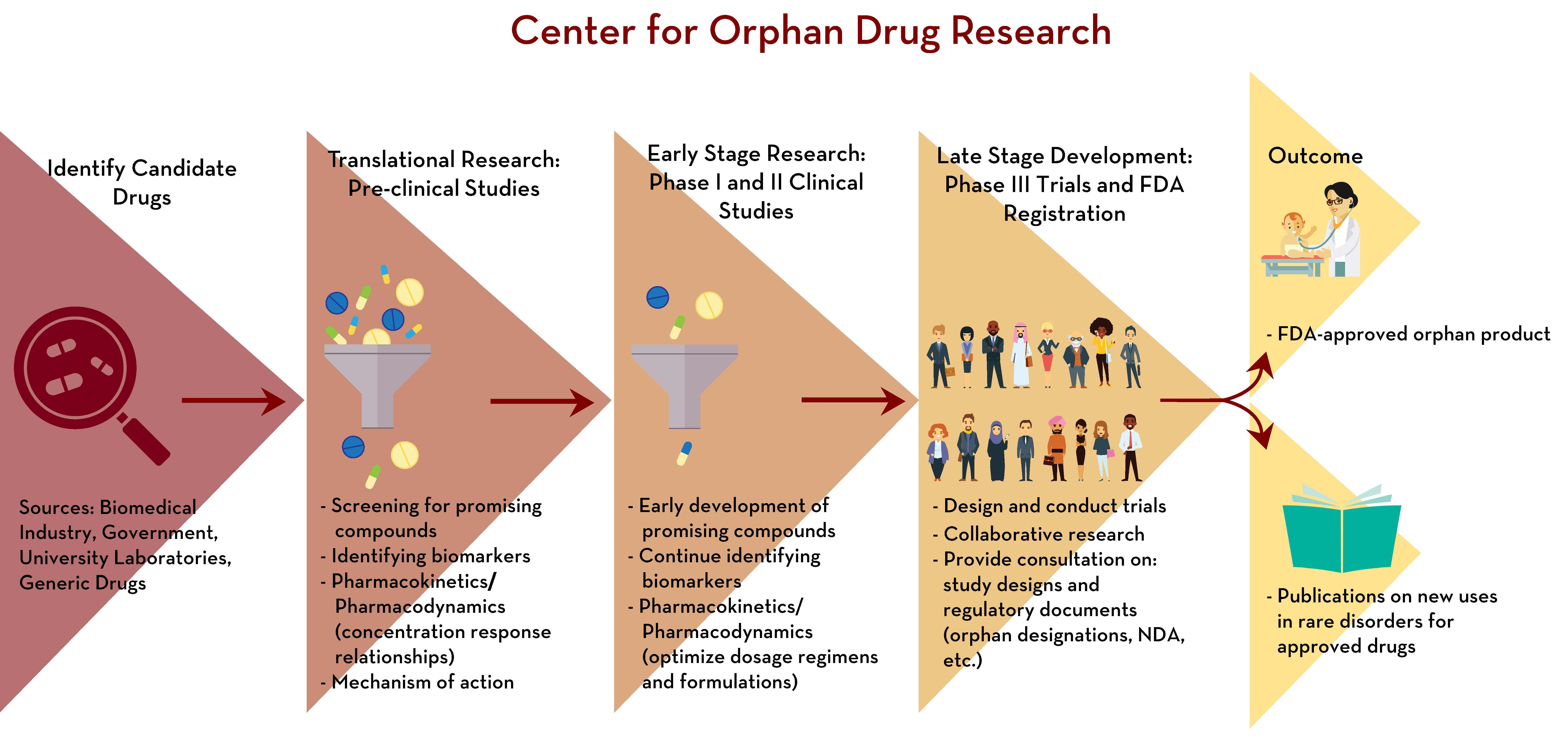
02/27/2021: Johnson & Johnson COVID-19 Vaccine Authorized by U.S. FDA For Emergency Use - First Single-Shot Vaccine in Fight Against Global Pandemic
Continue reading...- Prev
- 1
- Next
Also check out R-bloggers for lots of cool R stuff!